Research Article
| Data mining crystallization databases: Knowledge-based approaches to optimize protein crystal screens |
Matthew S. Kimber 1, François Vallee 1, Simon Houston 1, Alexander Ne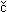 akov 1, Tatiana Skarina 2, Elena Evdokimova 2, Steven Beasley 2, Dinesh Christendat 2, Alexei Savchenko 2, Cheryl H. Arrowsmith 1 2, Masoud Vedadi 1, Mark Gerstein 3, Aled M. Edwards 1 2 * akov 1, Tatiana Skarina 2, Elena Evdokimova 2, Steven Beasley 2, Dinesh Christendat 2, Alexei Savchenko 2, Cheryl H. Arrowsmith 1 2, Masoud Vedadi 1, Mark Gerstein 3, Aled M. Edwards 1 2 * |
1Affinium Pharmaceuticals Inc., Toronto, Ontario, Canada
2Division
of Molecular and Structural Biology, Ontario Cancer Institute and
Department of Medical Biophysics, University of Toronto, Toronto,
Ontario, Canada
3Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut
|
| email: Aled M. Edwards (aled.edwards@utoronto.ca) |
*Correspondence
to Aled M. Edwards, Affinium Pharmaceuticals Inc., 10th floor, South
Tower, 100 University Avenue, Toronto, Ontario M5J 1V6, Canada
Funded by:
 Ontario Research and Development Challenge Fund
Ontario Research and Development Challenge Fund
Best Foundation
| crystal screening • crystallization • data mining • structural proteomics |
| Protein
crystallization is a major bottleneck in protein X-ray crystallography,
the workhorse of most structural proteomics projects. Because the
principles that govern protein crystallization are too poorly
understood to allow them to be used in a strongly predictive sense, the
most common crystallization strategy entails screening a wide variety
of solution conditions to identify the small subset that will support
crystal nucleation and growth. We tested the hypothesis that more
efficient crystallization strategies could be formulated by extracting
useful patterns and correlations from the large data sets of
crystallization trials created in structural proteomics projects. A
database of crystallization conditions was constructed for 755
different proteins purified and crystallized under uniform conditions.
Forty-five percent of the proteins formed crystals. Data mining
identified the conditions that crystallize the most proteins, revealed
that many conditions are highly correlated in their behavior, and
showed that the crystallization success rate is markedly dependent on
the organism from which proteins derive. Of the proteins that
crystallized in a 48-condition experiment, 60% could be crystallized in
as few as 6 conditions and 94% in 24 conditions. Consideration of the
full range of information coming from crystal screening trials allows
one to design screens that are maximally productive while consuming
minimal resources, and also suggests further useful conditions for
extending existing screens. Proteins 2003;51:562-568. © 2003
Wiley-Liss, Inc. |
Received: 2 April 2002; Accepted: 13 September 2002
10.1002/prot.10340 About DOI
INTRODUCTION
The
ultimate goal of structural proteomics is to obtain, through
experimental or computational methods, 3D structural models for every
protein in nature. Driving this ambitious program is the expectation
that structural information will provide functional insights for many
of the proteins predicted by genome-sequencing efforts that cannot be
ascribed a function using current sequence homology-based approaches.
The challenges in structural proteomics are significant; it has been
estimated that some 16,000 structures will have to be determined using
experimental approaches to obtain reasonable coverage of fold space.[1]
Recent advances in X-ray crystallography methodology and associated
technologies have made it possible, at least in ideal cases, to go from
data collection to a refined structure in a matter of hours[2-6]; however, actually growing a diffraction-quality crystal is far more time and resource intensive.
The
purpose of protein crystallization trials is to efficiently find useful
lead conditions from which crystal size and morphology can be
optimized. Commonly, one explores a wide range of different solutions
in which the salt concentration and type, pH, additive type,
temperature, and precipitant type and concentration are varied. The
precipitant is usually a long-chain polymer [poly-ethylene glycol
(PEG), jeffamine], an inorganic or organic salt, or some small organic
molecule (MPD, isopropanol, ethanol).[2]
There
are two general approaches to the design of crystallization screens.
One strategy aims to blanket potentially useful crystallization space
with screens of a few hundreds to over a thousand conditions (see,
e.g., the JBScreen at www.jenabioscience.com/jbscreen.html). The other strategy is to use smaller, more efficient screens based on previously successful conditions.[3-8]
The most widely used variant of this second strategy, developed by
Jancarik and Kim, is based on an incomplete factorial approach, which
explores a range of conditions biased toward previously successful
crystallization conditions.[8] The popularity of
this screening strategy can be ascribed to many reasons, including its
economy (1-2 mg of protein are needed), ease of use, manageable size,
and convenience (it is sold in preformulated kit form by various
companies). As originally formulated, this screen was intended to be
modified reiteratively as the experiences of users were incorporated.[3]
In practice, however, this has not happened. Partly this is due to a
lack of a clear metric by which to decide which of a set of potential
screening conditions is better, but also, more fundamentally, a lack of
sufficient, standardized experimental data by which to evaluate a
screen. While the optimal conditions for crystallizing a particular
protein crystal form are often collected and archived in a database,[9]
the detailed results of each screen, including partial successes or
failures, are not. As a result, while individual crystallization
conditions can perhaps be shown to be more or less successful,
researchers have tended to supplement the common factorial screens with
additional screens rather than attempting to systematically optimize
any given one. This may be a reasonable strategy when trying to
crystallize one or two proteins, in which the effort and expense
involved in making the additional protein, setting up the screen, and
evaluating the results is manageable. However, in the context of
structural proteomics this extra effort and expense is multiplied over
hundreds or even thousands of proteins, and thus the desirability of
screens that minimize the number of experiments while maximizing the
probability of success becomes far more pronounced. Here, by collating
data collected for 755 proteins from 6 organisms we show that it is
possible to use the information gleaned from previous screens to
improve screening strategies using objective empirical criteria.
METHODS
Proteins predicted not to have membrane-spanning domains from six organisms - the prokaryotes Staphylococcus aureus, Escherichia coli K12, Pseudomonas aeruginosa, Heliobacter pylori, and Thermotoga maritima and the archaeote Methanobacterium thermoautotrophicum - were amplified by PCR, cloned into E. coli expression vectors, overexpressed, and purified using His6 technology, as described elsewhere (see [[10]] for a general overview and [[11][12]]
for typical examples of procedures). Proteins were typically stored at
4°C, in 20 mM HEPES, pH 7.5, and 500 mM NaCl. The solutions for the
initial screen were purchased from Hampton Research. Proteins were
screened in 24-well Lindbro plates, using a 2-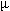 l + 2-l drop size and 700 l
in the well. For some proteins both the his-tagged and non-his-tagged
sample were both screened, and these were scored as separate samples.
Samples also included separate domains of multidomain proteins. For
most samples, two to four protein concentrations, typically ranging
from 5-40 mg ml-1, were screened in parallel; in almost all
cases this included at least one experiment in the 10- to 15-mg/ml
range. The data for these multiple experiments were pooled. All
crystallization experiments were performed at ambient temperature
(approximately 293 K). In total, over 35,000 experiments were performed.
l + 2-l drop size and 700 l
in the well. For some proteins both the his-tagged and non-his-tagged
sample were both screened, and these were scored as separate samples.
Samples also included separate domains of multidomain proteins. For
most samples, two to four protein concentrations, typically ranging
from 5-40 mg ml-1, were screened in parallel; in almost all
cases this included at least one experiment in the 10- to 15-mg/ml
range. The data for these multiple experiments were pooled. All
crystallization experiments were performed at ambient temperature
(approximately 293 K). In total, over 35,000 experiments were performed.
Screening
results were scored by eye after approximately 1 day, 1 week, 1 month,
and 3 months. For proteins where screening multiple samples at
different protein concentrations yielded different outcomes, only the
most favorable outcome was scored and reported. To minimize
subjectivity in scoring, results for each experiment were reduced to
one of three assessments - clear, precipitate, or crystalline. Samples
were required to have at least two conditions in which they are soluble
and two where they are not and no more than five conditions for which
data was missing (if, e.g., the condition was not set or the drop dried
out before it could be scored). These criteria reject 6 E. coli proteins, 3 T. martima proteins, 6 M. thermoautotrophicum proteins, and 55 S. aureus proteins. Minimal screen 6 was derived by sequentially searching all combinations of conditions (48!/6! 42! 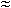 1.2 × 107
combinations) for the one that crystallized the most proteins. Minimal
screen 12 was derived by using minimal screen 6 as a seed and searching
all combinations of the remaining conditions for the six that best
complemented minimal screen 6. Minimal screen 24 was found by repeating
this condition twice more. Although this procedure is not guaranteed to
find the globally optimal subsets, this limitation is likely far less
serious than the one imposed by the limited amount of data available.
Clustering was performed in ClustalX using pairwise identity scores
once screening results had been
1.2 × 107
combinations) for the one that crystallized the most proteins. Minimal
screen 12 was derived by using minimal screen 6 as a seed and searching
all combinations of the remaining conditions for the six that best
complemented minimal screen 6. Minimal screen 24 was found by repeating
this condition twice more. Although this procedure is not guaranteed to
find the globally optimal subsets, this limitation is likely far less
serious than the one imposed by the limited amount of data available.
Clustering was performed in ClustalX using pairwise identity scores
once screening results had been  encoded
encoded as amino acid sequences.[13] Cladogram was produced in Phylodraw.[14]
as amino acid sequences.[13] Cladogram was produced in Phylodraw.[14]
RESULTS AND DISCUSSION
A total of 755 protein samples from T. maritima, E. coli, M. thermoautotrophicum, S. aureus, P. aeruginosa, and H. pylori were screened against conditions 1-48 of the Jancarik and Kim screen. The success rates for different genomes (Table I) ranged from 67.6% (for T. maritima, albeit with the smallest sample size) to 36.7% in the case of H. pylori.
The differences may reflect differences in the intrinsic properties of
proteins from different organisms, perhaps influenced by the
intracellular environment of the natural host, or may simply reflect
the quality of the protein produced by the E. coli host.
Overall, 338 protein samples (45%) yielded at least 1 crystallization
lead. For each protein that crystallized, crystals were observed, on
average, in 4.7/48 conditions.
| |
| Table I. Breakdown of Results by Source Organism |
|
| Organism | Number of proteins screened | Number of proteins crystallized | Mean hits per protein crystallizeda | % success |
|---|
| | S. aureus | 372 | 142 | 4.2 | 38.2 | | H. pylori | 128 | 47 | 6.4 | 36.7 | | E. coli | 116 | 72 | 4.8 | 62.1 | | M. thermoautotrophicum | 95 | 41 | 4.8 | 43.2 | | Th. maritima | 34 | 23 | 3.5 | 67.6 | | P. aeruginosa | 21 | 13 | 6.0 | 61.9 | | Overall | 755 | 338 | 4.7 | 44.7 |
|
|
a The
average number of conditions under which crystals were obtained,
considering only those samples for which at least one crystal was
obtained.
|
There were large differences in the number of proteins that crystallized in each condition (Table II),
ranging from 76 for condition 9 to 4 for condition 27. Surprisingly,
for 99 of the 338 proteins crystallized (29.3%) crystals were obtained
in only one condition; this is approximately 10-fold higher than might
be expected if crystallization in different conditions were behaving as
independent random variables (48 × 0.1 × 0.947 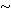 3.4%).
3.4%).
| |
Table II. Overview of Results Produced by the Jancarik and Kim Screen |
|
| Jancarik-Kim no. | Components of screening condition | pHa | Total clear | Total precipitate | Total crystals | Only crystalb |
|---|
| | 1 | 30% MPD Na Acetate pH 4.6 0.02 M CaCl2 | 5.06 | 197 | 540 | 17 | 1 | | 2 | 0.4 M K,Na Tartrate | 7.27 | 609 | 138 | 8 | 0 | | 3 | 0.4 M NH4 Phosphate | 4.24 | 264 | 450 | 12 | 0 | | 4 | 2.0 M NH4 Sulfate Tris.HCl pH 8.5 | 8.31 | 208 | 494 | 50 | 3 | | 5 | 30% MPD Na Hepes pH 7.5 0.2 M Na Citrate | 7.44 | 346 | 396 | 11 | 0 | | 6 | 30% PEG 4000 Tris.HCl pH 8.5 0.2 M MgCl2 | 8.70 | 84 | 601 | 65 | 4 | | 7 | 1.4 M Na Acetate Na Cacodylate pH 6.5 | 6.83 | 513 | 211 | 27 | 0 | | 8 | 30% Isopropanol Na Cacodylate pH 6.5 0.2 M Na Citrate | 7.06 | 201 | 544 | 9 | 1 | | 9 | 30% PEG 4000 Na Citrate pH 5.6 0.2 M NH4 Acetate | 6.54 | 108 | 568 | 76 | 1 | | 10 | 30% PEG 4000 Na Acetate pH 4.6 0.2 M NH4 Acetate | 5.82 | 70 | 632 | 49 | 4 | | 11 | 1.0 M NH4 Phosphate Na Citrate pH 5.6 | 4.89 | 309 | 404 | 15 | 0 | | 12 | 30% Isopropanol Na Hepes pH 7.5 0.2 M MgCl2 | 7.29 | 177 | 560 | 16 | 1 | | 13 | 30% PEG 400 Tris.HCl pH 8.5 0.2 M Na Citrate | 8.84 | 551 | 193 | 10 | 2 | | 14 | 28% PEG 400 Na Hepes pH 7.5 0.2 M CaCl2 | 7.32 | 212 | 517 | 25 | 2 | | 15 | 30% PEG 8000 Na Cacodylate pH 6.5 0.2 M NH4 Sulfate | 6.68 | 123 | 568 | 60 | 1 | | 16 | 1.5 M Li Sulfate Na Hepes pH 7.5 | 7.68 | 437 | 288 | 30 | 1 | | 17 | 30% PEG 4000 Tris.HCl pH 8.5 0.2 M Li Sulfate | 8.96 | 134 | 544 | 70 | 3 | | 18 | 20% PEG 8000 Na Cacodylate pH 6.5 0.2 M Mg Acetate | 6.62 | 137 | 546 | 72 | 1 | | 19 | 30% Isopropanol Tris.HCl pH 8.5 0.2 M NH4 Acetate | 8.37 | 218 | 525 | 7 | 0 | | 20 | 25% PEG 4000 Na Acetate pH 4.6 0.2 M NH4 Sulfate | 4.95 | 56 | 658 | 37 | 1 | | 21 | 30% MPD Na Cacodylate pH 6.5 0.2 M Mg Acetate | 6.71 | 265 | 468 | 19 | 3 | | 22 | 30% PEG 4000 Tris.HCl pH 8.5 0.2 M Na Acetate | 8.96 | 95 | 590 | 65 | 2 | | 23 | 30% PEG 400 Na Hepes pH 7.5 0.2 M MgCl2 | 7.28 | 276 | 454 | 25 | 1 | | 24 | 20% Isopropanol Na Acetate pH 4.6 0.2 M CaCl2 | 4.64 | 159 | 584 | 11 | 0 | | 25 | 1.0 M Na Acetate, Imidazole pH 6.5 | 7.90 | 566 | 166 | 18 | 1 | | 26 | 30% MPD Na Citrate pH 5.6 0.2 M NH4 Acetate | 6.50 | 235 | 509 | 7 | 1 | | 27 | 20% Isopropanol Na Hepes pH 7.5 0.2 M Na Citrate | 7.48 | 280 | 470 | 4 | 2 | | 28 | 30% PEG 8000 Na Cacodylate pH 6.5 0.2 M Na Acetate | 6.89 | 87 | 602 | 65 | 2 | | 29 | 1.6 M K,Na Tartrate Na Hepes pH 7.5 | 7.67 | 554 | 180 | 16 | 0 | | 30 | 30% PEG 8000 0.2 M NH4 Sulfate | 3.84 | 39 | 676 | 38 | 3 | | 31 | 30% PEG 4000 0.2 M NH4 Sulfate | 3.78 | 147 | 572 | 35 | 0 | | 32 | 2.0 M NH4 Sulfate | 5.01 | 201 | 517 | 36 | 2 | | 33 | 4.0 M Na Formate | 7.68 | 328 | 387 | 36 | 3 | | 34 | 2.0 M Na Formate Na Acetate pH 4.6 | 5.48 | 274 | 444 | 34 | 2 | | 35 | 1.6 M K,Na Phosphate Na Hepes pH 7.5 | 4.52 | 241 | 445 | 15 | 2 | | 36 | 8% PEG 8000 Tris.HCl pH 8.5 | 8.61 | 371 | 359 | 22 | 5 | | 37 | 8% PEG 4000 Na Acetate pH 4.6 | 4.85 | 192 | 536 | 24 | 1 | | 38 | 1.4 M Na Citrate Na Hepes pH 7.5 | 7.95 | 90 | 597 | 62 | 11 | | 39 | 2.0 M NH4 Sulfate Na Hepes pH 7.5 2% PEG 400 | 7.67 | 198 | 492 | 63 | 6 | | 40 | 20% Isopropanol + 20% PEG 4000 Na Citrate pH 5.6 | 6.59 | 157 | 565 | 30 | 0 | | 41 | 10% Isopropanol + 20% PEG 4000 Na Hepes pH 7.5 | 7.46 | 134 | 560 | 58 | 6 | | 42 | 20% PEG 8000 0.05 M K Phosphate | 4.62 | 89 | 586 | 52 | 3 | | 43 | 30% PEG 1500 | 5.36 | 101 | 594 | 54 | 7 | | 44 | 0.2 M Mg Formate | 6.86 | 473 | 253 | 26 | 1 | | 45 | 18% PEG 8000 Na Cacodylate pH 6.5 0.2 M Zn Acetate | 5.85 | 160 | 572 | 19 | 5 | | 46 | 18% PEG 8000 Na Cacodylate pH 6.5 0.2 M Ca Acetate | 6.50 | 120 | 570 | 57 | 2 | | 47 | 2.0 M NH4 Sulfate Na Acetate pH 4.6 | 4.64 | 100 | 619 | 30 | 1 | | 48 | 2.0 M NH4 Phosphate Tris.HCl pH 8.5 | 4.25 | 216 | 461 | 14 | 1 | | Totals | | | 11,102 | 23,205 | 1601 | 99 |
|
|
Buffers when present were at 0.1 M.
a pH is experimentally measured pH, performed in duplicate on two different batches of the screen.
b Number of proteins for which this condition yielded the only crystal lead.
|
Efficacy of Different Precipitants
In
17 of the 48 conditions, salt was the major precipitant. These
conditions together crystallized a total of 200 of the 338 proteins
(59.2%) [Fig. 1(a)]. There were significant
differences in the effectiveness of different salts. Sodium citrate,
for example, is represented by a single condition in the screen, 39,
but was the eighth most productive condition in the screen. Moreover,
this condition was by far the most likely condition to yield the only
crystal for a given protein. This unusual behavior may be related to
citrate's strong metal chelating abilities, something that suggests
further investigation.[2] Ammonium sulphate, long
considered an exceptionally good salt for crystallizing proteins, was
the major precipitant in four conditions (4, 32, 39, and 47), all of
which yield substantial numbers (50, 36, 63, and 30) of crystals.
Lithium sulphate (16) and formate salts (33, 34, and 44) were also
successful. Citrate, sulphate, and formate salts formed a cluster that
affected protein solubility in a similar fashion; they tended to
crystallize the same subset of proteins (Fig. 2).
The other salts used, namely, phosphate (3, 11, 35, and 48), acetate (7
and 25), and tartrate (2 and 29), were much less successful
precipitants; in the case of phosphate, this likely reflected the fact
that all of these conditions are acidic. Only 69 proteins crystallized
in these 8 conditions.
 | Figure 1. (a)
Venn diagram showing the number of proteins for which crystals were
obtained in conditions where salt (17 screening conditions, red), PEG
(22 conditions, blue), or an organic molecule (9 conditions, green) was
the major precipitant. (b) Number of proteins with a given
number of successful screening conditions. While some exceptional
samples crystallize in up to two-thirds of all conditions, most
proteins crystallize in relatively few conditions. (c) Number of
crystals contained for a given number of selected screening conditions.
Conditions were added one at a time, where the condition added was the
one that most increased the number of crystals obtained relative to the
previously chosen subset. Note that almost all of the crystals obtained
can be obtained from approximately half of the screening conditions and
that the last nine conditions could be omitted without affecting the
number of samples crystallized.
[Normal View 32K | Magnified View 106K] |
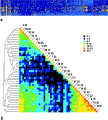 | Figure 2. (a)
Subset of the overall data, representing the 338 proteins successfully
crystallized. Different conditions are arrayed vertically and different
proteins horizontally. Yellow squares represent crystals, dark blue
squares represent precipitated proteins, and cyan squares represent
soluble proteins. Proteins and conditions were sequentially clustered
using ClustalX using identity matrices for scoring. (b) Distance
matrix of clustered conditions. Condition numbers are noted along the
diagonal; off-diagonal elements represent the number of instances in
which proteins were found to crystallize in both the condition to the
right and the one above it. Note the strong correlation between the PEG
conditions (bottom right corner) as well as the
citrate/sulphate/formate salts (top left corner). The scale is
logarithmic. On the left is a Cladogram
showing the degree of relatedness of crystal screen conditions as
inferred from the degree to which they crystallize the same protein
samples. Note that the crystallization conditions cluster primarily on
the basis of the chemical nature of the major precipitant and
secondarily on the basis of pH.
[Normal View 40K | Magnified View 147K] |
High-molecular-weight PEGs are widely considered the most successful protein crystallization agents.[2][9]
Of the 338 total proteins, 229 crystallized (67.7%) in the 14
conditions that consist of PEG 4000 or 8000 at concentrations greater
than 18%, possibly with some buffer and inert salt (excluding condition
45, which contains zinc), for an average of 54 crystals per condition.
The six most productive conditions overall were all in this group.
However, there was, in general, a great deal of redundancy among the
PEG conditions [Fig. 2(b)]. This region of
parameter space appears to be heavily oversampled, a consequence of
strong bias toward previously successful experiments built into the
original design of the screen. Low concentrations of PEG (36, 37)
appeared to be less effective at obtaining crystals but crystallized a
different set of proteins than those obtained at higher concentrations.
PEG 400 (13, 14, and 23) appeared to affect the solubility of proteins
in a manner that more resembled the action of a salt than that of a
high-molecular-weight PEG. This may reflect the fact that smaller PEGs
probably precipitate proteins more by a solvent competition effect than
a volume exclusion effect. Also, zinc in conjunction with PEG yielded
different solubility behavior than other salt/PEG combinations, likely
reflecting the ability of transition metals to effect strong
interactions between proteins by virtue of their ligand chemistry.
Organic
precipitants formed a minor part of the screen. There are four MPD
conditions (1, 5, 21, and 26) that yielded a modest numbers of crystals
(39 proteins crystallized, average of 14 crystals per condition). The 5
conditions in which isopropanol was the primary precipitant (8, 12, 19,
24, and 27) yielded few crystals (average 9 crystals per condition, 39
proteins crystallized) unless combined with 20% PEG (40 and 41). In
vapor diffusion experiments, the slow diffusion of water from the drop,
where vapor pressure is higher, to the well, where vapor pressure is
lower, drives a gradual increase in protein and precipitant
concentration in the drop that may eventually lead to the
crystallization of the protein. Volatile precipitants, on the other
hand, tend to diffuse in the opposite direction, decreasing protein
concentration. In the case of isopropanol this happens rapidly, making
this precipitant perhaps better suited to batch experiments than vapor
diffusion. Overall, the 9 conditions that contain organic precipitants
crystallized 66 proteins, 10 of which grew only in these conditions.
Mining the Information to Identify Minimal Screens
The
strong interdependence of various conditions of the Jancarik and Kim
screen implies that it is, in its present formulation, less than ideal.
Several conditions produced few crystals, while other groups of
conditions, such as those based on high-molecular-weight PEGs, were too
highly correlated. We set out to identify reduced condition sets that
minimally compromise the chances of getting at least one crystal.
Sequential searches of all combinations of conditions yielded a series
of minimal screens (Table III) that comprised sets of conditions optimized for maximal probability of successfully obtaining a crystal [Fig. 1(c)].
A potential drawback of optimizing coverage at the expense of
redundancy was that alternate crystal forms with different diffraction
qualities might be missed; in those instances where a different crystal
form would prove desirable, a second round of screening could then be
initiated.
| |
| Table III. Minimal Screens |
|
| Screen | Conditions included | # proteins crystallized | % crystals vs. full screen | % of total proteins crystallized |
|---|
| | Minimal screen 6 | 6, 10, 18, 38, 39, 43 | 205 | 61 | 27.1 | | Minimal screen 12 | 4, 6, 10, 17, 18, 30, 36, 38, 39, 41, 43, 45 | 268 | 79 | 35.5 | | Minimal screen 24 | 1, 4, 6, 10, 11, 13, 14, 16, 17, 18, 20, 21, 28, 30, 33, 34, 35, 36, 38, 39, 41, 42, 43, 45 | 318 | 94 | 42.1 | | JK 1-48 | 1-48 | 338 | 100 | 44.8 |
|
|
These
screens are not only potentially useful in their own right (e.g., in
cases where material is limited) but also may potentially serve as the
nucleus of a new, more efficient screen.
|
Six
conditions - 6, 10, 18, 38, 39, and 43 (referred to hereafter as
minimal screen 6) - yielded crystals for 205 of the 338 proteins
(60.6%) successfully crystallized by the full screen. It is interesting
to note the dispersion of these conditions - high-molecular-weight PEG
at acid, neutral, and basic pH (10, 18, and 6), low-molecular-weight
PEG (43), and two different salts (38 and 39). Augmenting this set with
conditions 4, 17, 30, 36, 41, and 45 (minimal screen 12) yielded 268 of
the 338 crystals (79.3%), and adding a further 12 conditions - 1, 11,
13, 14, 16, 20, 21, 28, 33, 34, 35, and 42 (minimal screen
24) - yielded 318 of the 338 crystals (94.1%). In addition to the
conditions defined as minimal screen 24, the omission of conditions 22,
23, 27, 32, and 46 from the screen would have each resulted in 2 fewer
proteins being crystallized, while the omission of conditions 8, 9, 12,
15, 25, 26, 37, 44, 47, and 48 would have each resulted in 1 fewer
protein being crystallized. Nine conditions - 2, 3, 5, 7, 19, 24, 29,
31, and 40 - could have been omitted from the screen entirely without
losing a single crystal from 755 samples.
Note that had the
conditions for the minimal screens been chosen by considering only the
total number of crystals produced per condition significantly less
productive screens would have resulted. For example, the 6 individually most productive conditions crystallized 180 proteins compared to 205 for the optimal 6.
Screening
data can also be mined for trends in precipitation. For example, the
conditions employing tartrate and acetate salts as the primary
precipitant were not only among the poorest crystal producers but also
among those least likely to precipitate a protein. Because for the
majority of proteins supersaturation was never reached with these
precipitants, a substantially higher concentration may be predicted to
be more effective.
Rational Strategy for Producing Maximally Productive Screens
Although
the Jancarik and Kim screen has proved a useful tool for a generation
of crystallographers, it is clear that a more efficient screen of a
similar size could be derived from it by substituting some of its less
productive conditions with ones chosen that demonstrably complement its
more productive conditions. With sufficient data it is a relatively
straightforward procedure to eliminate those conditions that contribute
little, and iterative cycles of further additions, testing, and
elimination should allow the eventual optimization of the screen.
Analysis of the data generated may also help suggest suitable candidate
conditions for expanding the screen. Conditions that show a strong
tendency to uniquely crystallize proteins are likely in regions of
parameter space that are undersampled and could therefore yield more
crystals. For example, of the 62 proteins crystallized by condition 38,
11 are uniquely crystallized by this condition, implying that citrate
salts have some unique properties whose potential for crystallization
is underexploited by the present screen. Similarly, further conditions
with transition metal ions (such as condition 45) and
intermediate-molecular-weight PEGs (such as in condition 43) might also
prove useful additions.
Employing such a strategy, one can
experiment with a wide variety of conditions without increasing the
amount of work to unmanageable proportions and without sacrificing the
proven productivity of a core set of conditions - important
considerations given that the only practical manner to obtain
sufficient samples and data to implement this strategy is to
incorporate it into ongoing structural proteomics efforts. Also,
because of the tentative nature of additions, this strategy should
encourage the exploration of chemically diverse conditions that might
otherwise not be thought sufficiently safe to include in a fixed, generally used screen.
In
this study, a crystallization database was created and mined to find
the most productive screening conditions, highlighting the value of
such efforts within structural proteomics projects. Clearly, not only
this sort of information can be extracted from the data, and there is
more analysis to perform. For example, whereas this data set comprises
the best conditions to identify initial crystals, most of these
crystals have not been optimized to form diffraction-quality crystals.
It will be interesting to learn whether there are differences in the
suitability of different conditions as a starting point for producing
large, well-diffracting crystals. The data could also be analyzed for
solubility properties rather than crystallization. In this way, one may
be able to identify a set of solution conditions that most often yield
soluble protein - something potentially useful for NMR experiments, for
example. Both solubility data and crystallization data should
ultimately be linked to the biophysical properties of the proteins,
such as the isoelectric point or amino acid content. Clearly, the
crystallization databases resulting from structural proteomics projects
can yield important information, and efforts should be expended to
ensure that they are routinely collected in a consistent,
machine-readable format.
CONCLUSIONS
Initial
crystallization conditions for a novel macromolecular sample are
obtained by screening the sample against a wide variety of chemical cocktails,
in general a generic set preselected for their historically proven
efficacy in producing crystals. Despite the critical nature of this
step, however, no systematic effort has been made to optimize the set
of conditions to be used. Here, we used data generated by subjecting a
large set of proteins against a commonly used, commercially available
screen to obtain a clearer picture of the overall efficacy of the
present screening strategies and see if there are obvious ways to
improve them. This data leads to several nontrivial conclusions: (1)
Among archaeal and bacterial genomes, there appear to be large
differences in the degree to which proteins are tractable to
crystallization; (2) a small subset of the conditions, even in the
relatively small Jancarik and Kim screen, are responsible for a large
proportion of crystals obtained overall; (3) as a corollary to this,
screening hundreds of conditions, as advocated in some screening
protocols, is little more likely to yield a crystal than searching a
few tens of well-chosen conditions. The results of this experiment
suggest that iteratively adding new conditions, testing against a large
set of proteins, and rejecting those conditions that contribute least
should allow the fine-tuning of existing screens while still having a
useful screen in place at all times. Ultimately this will be of great
benefit to structural proteomics efforts as an efficient, optimized
screen will give maximal samples for structure solution while
minimizing the amount of time and material wasted on unneeded
experiments.
Acknowledgements
This
work was supported in part by the Ontario Research and Development
Challenge Fund. A.M.E. and C.H.A. are CIHR Investigators. D.C. was
supported by a fellowship from the Best Foundation.
| 1 |
Vitkup D,
Melamud E,
Moult J,
Sander C.
Completeness in structural genomics.
Nat Struct Biol
2001;
8:
559-566. Links |
| 2 |
McPherson A.
Crystallization of Biological Macromolecules.
Cold Spring Harbor, ME:
Cold Spring Harbor Laboratory Press;
1999. p.
586. |
| 3 |
Jankarik J,
Kim SH.
Sparse matrix sampling: a screening method for crystallization of proteins.
J Appl Crystallogr
1991;
24:
409-411. Links |
| 4 |
Kingston R,
Baker H,
Baker E.
Search designs for protein crystallization based on orthogonal arrays.
Acta Crystallogr D
1994;
50:
429-440. Links |
| 5 |
Saridakis E,
Chayen N.
Improving protein crystal quality by decoupling nucleation and growth in vapor diffusion.
Protein Sci
2000;
9:
755-757. Links |
| 6 |
Scott W,
Finch J,
Grenfell R,
Fogg J,
Smith T,
Gait M,
Klug A.
Rapid crystallization of chemically synthesized hammerhead RNAs using a double screening procedure.
J Mol Biol
1995;
250:
327-332. Links |
| 7 |
Cudney B,
Patel S,
Weisgraber K,
Newhouse Y,
McPherson A.
Screening and optimization strategies for macromolecular crystal growth.
Acta Crystallogr D
1994;
50:
414-423. Links |
| 8 |
Carter CJ,
Carter C.
Protein crystallization using incomplete factorial experiments.
J Biol Chem
1979;
254:
12219-12223. Links |
| 9 |
Gilliland G, Tung M, Blakeslee D, Ladner J. Biological Macromolecule
Crystallization Database, Version 3.0: new features, data and the NASA
archive for protein crystal growth data. Acta Crystallogr D
1994;
50:
408-413. Links |
| 10 |
Christendat D,
Yee A,
Dharamsi A,
Kluger Y,
Savchenko A,
Cort JR,
Booth V,
Mackereth CD,
Saridakis V,
Ekiel I,
Kozlov G,
Maxwell KL,
Wu N,
McIntosh LP,
Gehring K,
Kennedy MA,
Davidson AR,
Pai EF,
Gerstein M,
Edwards AM,
Arrowsmith CH.
Structural proteomics of an archaeon.
Nat Struct Biol
2000;
7:
903-909. Links |
| 11 |
Wu N,
Christendat D,
Dharamsi A,
Pai EF.
Purification, crystallization and preliminary X-ray study of orotidine 5 -monophosphate decarboxylase.
Acta Crystallogr D Biol Crystallogr
2000;
56
(7):
912-914. Links -monophosphate decarboxylase.
Acta Crystallogr D Biol Crystallogr
2000;
56
(7):
912-914. Links |
| 12 |
Zhang R,
Skarina T,
Katz J,
Beasley S,
Khachatryan A,
Vyas S,
Arrowsmith C,
Clarke S,
Edwards A,
Joachimiak A,
Savchenko A.
Structure of Thermotoga maritima stationary phase survival protein SurE: a novel acid phosphatase.
Structure
2001;
9:
1095-1106. Links |
| 13 |
Jeanmougin F,
Thompson J,
Gouy M,
Higgins D,
Gibson T.
Multiple sequence alignment with Clustal X.
Trends Biochem Sci
1998;
23:
403-405. Links |
| 14 |
Choi J,
Jung H,
Kim H,
Cho H.
PhyloDraw: a phylogenetic tree drawing system.
Bioinformatics
2000;
16:
1056-1058. Links |
Copyright © 1999-2003 by John Wiley & Sons, Inc. All rights reserved.