A Database of Macromolecular Motions
Mark Gerstein
&
Werner Krebs
Department of Molecular Biophysics & Biochemistry
266 Whitney Avenue, Yale University
PO Box 208114, New Haven, CT 06520
(203) 432-6105, FAX (203) 432-5175
Mark.Gerstein@yale.edu
Keywords: Structure Databank, Protein Structure, Biophysics, Protein Folding
Running Title: A Database of Macromolecular Motions
Manuscript is 25 Pages in Length (including this one)
Version: mm715
Abstract
We describe a database of macromolecular motions meant to be of general use to the structural community. The database, which is accessible on the World Wide Web with an entry point at http://bioinfo.mbb.yale.edu/MolMovDB, attempts to systematize all instances of protein and nucleic acid movement for which there is at least some structural information. At present it contains ~120 motions, most of which are of proteins. Protein motions are further classified hierarchically into a limited number of categories, first on the basis of size (distinguishing between fragment, domain, and subunit motions) and then on the basis of packing. Our packing classification divides motions into various categories (shear, hinge, other) depending on whether or not they involve sliding over a continuously maintained and tightly packed interface. In addition, the database provides some indication about the evidence behind each motion (i.e. the type of experimental information or whether the motion is inferred based on structural similarity) and attempts to describe many aspects of a motion in terms of a standardized nomenclature (e.g. the maximum rotation, the residue selection of a fixed core, etc). Currently, we use a standard relational design to implement the database. However, the complexity and heterogeneity of the information kept in the database makes it an ideal application for an object-relational approach, and we are moving it in this direction. Specifically, in terms of storing complex information, the database contains plausible representations for motion pathways, derived from restrained 3D interpolation between known endpoint conformations. These pathways can be viewed in a variety of movie formats, and the database is associated with a server that can automatically generate these movies from submitted coordinates.
Introduction
Motions of macromolecules (proteins and nucleic acids) are often the essential link between structure and function; that is, motion is frequently the way a structure actually carries out a particular function. Protein motions, in particular, are involved in many basic functions such as catalysis, regulation of activity, transport of metabolites, formation of large assemblies and cellular locomotion. Highly mobile proteins have, in fact, been implicated in a number of diseases -- e.g., the motion of gp41 in AIDS and that of the prion protein in scrapie (19, 27, 45, 79, 111).
Macromolecular motions are also of intrinsic interest because of their fundamental relationship to the principles of protein and nucleic acid structure and stability. They are, however, among the most complicated biological phenomena that can be studied in great quantitative detail, involving concerted changes in thousands of precisely specified atomic coordinates. Moreover, the time scales of macromolecular motions range over more than nine orders of magnitude (from sub-nanosecond loop closures to more than one second refoldings (26, 71, 74)) placing their study beyond any single type of experimental technique or numerical simulation.
Fortunately, it is now possible to study these motions in a database framework, by analyzing and systematizing many of the instances of protein structures solved in multiple conformations. We present here a comprehensive database of macromolecular motions, intended to be of use to those studying structure-function relationships (e.g. as in rational drug design (64)) and also to those involved in large-scale proteome or genome surveys (33, 37, 59). There are a number of reasons why it is favorable (and feasible) at present to construct such a database: (i) The amount of raw data (known protein and nucleic acid structures and sequences homologous to them) is rapidly increasing (15, 48, 78), and an increasing fraction of new structures have non-trivial motions (see below). (ii) The graphical and interactive nature of a database is particularly well suited for presenting macromolecular motions, which are often difficult to represent on a static journal page. (ii) A loose infrastructure of federated databases has emerged in the structural community, allowing the motions database to connect to a variety of information sources (114) (see list in caption to Figure 1).
Only one previous attempt has been made at the systematic classification of protein motions. Boutonnet et al. (14) do not present a database but rather develop an automatic tool for classifying proteins. In indirectly related work, a data set of protein interfaces has also been developed (108).
Overall Organization of the Database
A public interface to the database exists on the World Wide Web at http://bioinfo.mbb.yale.edu/MolMovDB . Presently, this consists of a set of coupled hypertext pages with graphic images and a simple query box, though more sophisticated interfaces are planned in the future. As shown in Figure 1, using the database is straightforward. One may browse either by typing various search keywords into the main page or by navigating through an outline. Either way brings one to the entries. Thus far, the database has more than 120 entries, which refer to over 240 structures in the Protein Databank (PDB) (Table 2). (Further information about the public interface to the database is described in the caption to Figure 1 and at http://bioinfo.mbb.yale.edu/MolMovDB/linkhelp.txt.)
Unique Motion Identifier
Each entry is indexed by a unique motion identifier, rather than around individual proteins and nucleic acids. This is because a single macromolecule can have a number of motions and the same essential motion can be shared amongst different macromolecules (see below). (The motion identifier is a short string like "igelbow," which attempts to evoke some characteristic of the motion or protein in the mnemonic style of the SwissProt identifiers (7).)
Attributes of a Motion
In addition to the motion identifier, each entry has the following information:
(i) Classification. A classification number gives the place of a motion in the size and packing classification scheme for motions described below. In addition to its basic classification, a motion can also be annotated as being "similar-to" or "sharing-characteristics-with" a motion in a different protein or "part-of" or "containing" another motion in the same protein. For instance, the motions in all the different bacterial sugar binding proteins are similar to each other (98, 110), and the domain closure in aspartate carbamoyltransferase is clearly part of and driven by a larger allosteric transition, involving the motion of subunits (103, 104).
(ii) Structures. Databank identifiers are given for the various conformations of the macromolecule (e.g. open and closed). These act as foreign keys into other databases. In particular, they have been used to link directly to the entries in the main protein and nucleic acid databases (PDB and NDB), to sequence and journal cross-references via the Entrez and MMDB, and to related structures via the Structural Classification of Proteins (SCOP) (3, 11, 28, 46, 51, 75, 96). In the more highly annotated entries, residue selections are given for the main rigid core, for other secondary cores moving rigidly relative to the main core, and for flexible hinge regions linking the cores.
(iii) Literature. Literature references are given. Where possible these are via Medline unique identifiers, allowing a link to be made into the PubMed database (28, 96).
(iv) Blurb. Each entry has a paragraph or so of plain text documentation. While this is, in a sense, the least precisely defined field, it is the heart of each entry, describing the motion in intelligible prose and referring to figures, where appropriate. The rationale behind each motion's classification is discussed, at least implicitly, here.
(v) Standardized Nomenclature. For many entries we describe the overall motion using standardized numeric terminology, such as the maximum displacement (overall and of just backbone atoms) and the degree of rotation around the hinge. These statistics are summarized in Table 1. We also attempt to give the transformations (from ii) needed to optimally superimpose and orient each coordinate set to best see the motion (i.e. down screw-axis) and the selections of residues with large changes in torsion angles, packing efficiency, or neighbor contacts.
(vi) Graphics. Each entry has links to graphics and movies describing the motion, often depicting a plausible interpolated pathway (see below).
Hierarchical Classification Scheme based on Size then Packing
Size Classification: Fragment, Domain, Subunit
In the classification scheme currently in use, the most basic division is between proteins and nucleic acids. There are far fewer motion entries for nucleic acids than for proteins, reflecting the much larger number of known protein structures. Currently, the database includes the nucleic-acid motions evident from comparing various conformations of the known structures of catalytic RNAs and tRNAs (specifically, the Hammerhead ribozyme, the P4-P6 domain of the Group II intron, and Asp-tRNA (18, 81, 85, 91, 97)).
The classification scheme for proteins has a hierarchical layout shown in Figure 2. The first division is based on the size of the motion. Ranked in order of their size, protein movements fall into three categories: the motions of subunits, domains, and fragments smaller than domains.
Nearly all large proteins are built from domains, and domain motions, such as those observed in hexokinase or citrate synthase (10, 86), provide the most common examples of protein flexibility (9, 39, 53). The motion of fragments smaller than domains usually refers to the motion of surface loops, such as the ones in triose phosphate isomerase or lactate dehydrogenase, but it can also refer to the motion of secondary structures, such as of the helices in insulin (2, 24, 113). Often domain and fragment motions involve portions of the protein closing around a binding site, with a bound substrate stabilizing a closed conformation. They, consequently, provide a specific mechanism for induced-fit in protein recognition (61, 62). In enzymes this closure around a binding site has been analyzed in particular detail (6, 57, 58, 92, 106). It serves to position important chemical groups around the substrate, shielding it from water and preventing the escape of reaction intermediates.
Subunit motion is distinctly different from fragment or domain motion. It affects two large sections of polypeptide that are not covalently connected. It is often part of an allosteric transition and tied to regulation (29, 80). For instance, the relative motions of the subunits in the transport protein hemoglobin and the enzyme glycogen phosphorylase change the affinity with which these proteins bind to their primary substrates (30, 54).
Packing Classification: Hinge and Shear
We have systematized the motions of protein domains and smaller units on the basis of packing, using an expanded version of a scheme developed previously (39). This is because the tight packing of atoms inside of proteins provides a most fundamental constraint on protein structure (42, 44, 68, 87-89). It is usually impossible for an atom inside a protein to move much without colliding with a neighboring atom, unless there is a cavity or packing defect (49, 50).
Internal interfaces between different parts of a protein are packed very tightly (35, 38, 39). Furthermore, they are not smooth, but are formed from interdigitating sidechains. Common sense consideration of these aspects of interfaces places strong constraints on how a protein can move and still maintain its close packing. Specifically, maintaining packing throughout a motion implies that the sidechains at the interface must maintain their same relative orientation and pattern of inter-sidechain contacts in both conformations (e.g. open and closed).
These straightforward constraints on the types of motions that are possible at interfaces allow an individual movement within a protein to be described in terms of two basic mechanisms, shear and hinge, depending on whether or not it involves sliding over a continuously maintained interface (39) (Figure 2). A complete protein motion (which can contain many of these smaller "movements") can be built up from these basic mechanisms. For the database, a motion is classified as shear if it predominately contains shear movements and as hinge if it is predominately composed of hinge movements. More detail on the characteristics of the two types of motion follow.
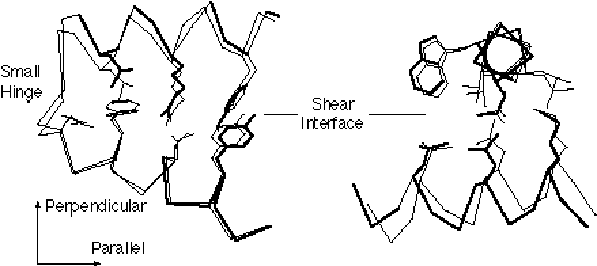
(i) Shear. As shown in figure 3, the shear mechanism basically describes the special kind of sliding motion a protein must undergo if it wants to maintain a well-packed interface. Because of the constraints on interface structure described above, individual shear motions have to be very small. Sidechain torsion angles maintain the same rotamer configuration (82) (with <15° rotation of sidechain torsions); there is no appreciable mainchain deformation; and the whole motion is parallel to the plane of the interface, limited to total translations of ~2 Å and rotations of 15°. Since an individual shear motion is so small, a single one is not sufficient to produce a large overall motion, and a number of shear motions have to be concatenated to give a large effect — in a similar fashion to each plate in a stack of plates sliding slightly to make the whole stack lean considerably. Consequently, proteins that undergo shear often have a layered architecture. Examples include citrate synthase, Trp repressor and aspartate amino transferase (39, 65, 66, 72).
(ii) Hinge. As shown in figure 4, hinge motions occur when there is no continuously maintained interface constraining the motion. These motions usually occur in proteins that have two domains (or fragments) connected by linkers (i.e. hinges) that are relatively unconstrained by packing. A few large torsion angle changes in the hinges are sufficient to produce almost the whole motion. The rest of the protein rotates essentially as a rigid body, with the axis of the overall rotation passing through the hinges. The overall motion is always perpendicular to the plane of the interface (so the interface exists in one conformation but not in the other, as in the closing and opening of a book) and is identical to the local motion at the hinge. Examples include lactoferrin and tomato bushy stunt virus (TBSV) (5, 77).
Gerstein et al. (36, 38, 40) analyzed the hinged domain and loop motion in specific proteins (lactate dehydrogenase, adenylate kinase, lactoferrin). These studies emphasized how critical the packing at the base of a protein hinge is -- in the same sense that the "packing" at the base of an everyday door hinge determines whether or not the door can close). Protein hinges are special regions of mainchain in that they are exposed and have few packing constraints on them and are thus free to sharply kink (Figure 4). Most mainchain atoms, in contrast, are usually buried beneath layers of other atoms (usually sidechain atoms), precluding large torsion angle changes and hinge motions. Conversely, the presence of a hinge does not appear to be related to chain topology or secondary structure -- i.e. mobile hinges have been found in loops, sheets, and helices.
It is important to emphasize that most shear motions do, in fact, contain hinges (joining the various sliding parts) and that the existence of a hinge is not the salient difference between the two basic mechanisms -- rather it is the existence of a continuously maintained interface.
Other Classification
Most of the fragment and domain motions in the database fall within the hinge-shear classification. However, there are a number of exceptions, and we have created some special categories to deal with them.
(i) A special mechanism that is clearly neither hinge nor shear accounts for the motion. An example of this sort of motion is what occurs in the immunoglobulin ball-and-socket joint (67), where the motion involves sliding over a continuously maintained interface (like a shear motion) but because the interface is smooth and not interdigitating the motion can be large (like a hinge).
(ii) Motion involves a partial refolding of the protein. This usually results in dramatic changes in the overall structure. Examples where both endpoints are known include the motion in the serpins and influenza virus haemagglutinin (17, 102). Also, included in this category are order-to-disorder transitions (as when a DNA recognition domain becomes ordered upon binding DNA), protein domains that only become structured upon oligomerization (e.g. leucine zipper dimerization domain), and pro-enzymes that dramatically change shape upon cleavage.
(iii) Motion cannot yet be classified. An example of this is the beta-sheet deformations in the TATA-box binding protein (20, 56).
For the motions of subunits a different division is made (other than hinge or shear):
(i) Allosteric. Examples include hemoglobin and aspartate carbamoyltransferase (30, 103, 104).
(ii) Non-allosteric. Examples include the quaternary structure change in the BamHI endonuclease upon binding DNA (76).
(iii) Complex motions. Large protein motions which involve many subsidiary "sub-motions" (which in themselves can be classified as subunit or domain motions) are put into the category of complex motions. The lac repressor, which contains three distinct motions, provides a good example of this situation (25, 69). The first motion is an order-to-disorder transition that the headpiece domain undergoes when it binds DNA. A second motion involves a molecule binding between two other domains in the protein. This motion is essentially the same as the motion observed in another group of proteins, the bacterial periplasmic binding proteins (110). However, it is coupled to a further subunit rearrangement that changes the overall DNA binding affinity of the protein and consequently is termed an allosteric transition. Finally, a third motion involves another subunit motion (which is not linked to the allosteric transition) that allows the four reading head domains to bind sites on DNA with different spacing and curvature.
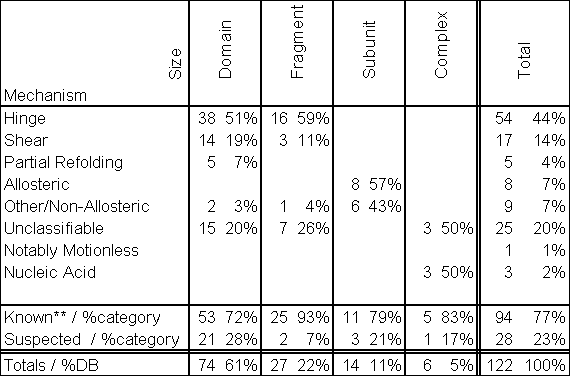
A breakdown of the categorization of entries in the current database is given in Table 2. At the time of this writing (version 1.71), the database describes 122 macromolecular motions which reference 249 PDB structures. The hinge mechanism is the most common classification in the database, accounting for 45% of the entries. Over 60% of the motions in the database are classified as domain motions. Interestingly, a greater percentage of fragment motions have structures for multiple conformations in the motion, probably reflecting the greater ease with which these smaller motions can be studied experimentally.
Annotation of Evidence related to the Motion
Levels of Annotation and Types of Experimental Information
For each entry in the database, we have tried to indicate the evidence behind its description and classification: i.e. is it based on careful manual analysis of two conformations, automatic output of a conformation comparison program, inferred based on structure comparison, or inferred based on sequence comparison? Thus, a clear distinction is made between the carefully documented, "gold-standard" motion in lactoferrin (i.e. as shown in Figure 4) and the much more tentatively understood motion in a protein that is a sequence homologue of another protein which is structurally similar to lactoferrin.
At present, nearly all entries in the motions database are the result of careful manual analysis and classification; thus, the current database is intended to serve as an accurate "core" around which a much larger, semi-automatically populated database may be constructed. We hope that this attention to the evidence behind the motion in the annotation will allow the database to grow rapidly in the future without becoming corrupted with false assertions.
Experimental information on macromolecular movements comes from a number of sources: X-ray structures of particular proteins and nucleic acids in different conformational states (typically "open" and "closed," but other configurations occur, e.g. in allostery and order-disorder transitions), NMR studies (e.g. Pf1 coat protein (99)), time-resolved studies (e.g. ras, PYP, bacteriorhodopsin (32, 94, 107)), fluorescence techniques, and small-angle scattering. There is much less information on the time scales of the motions in comparison to the detailed information on coordinate changes. Some 95% of entries in the database have been studied by traditional X-ray crystallography, and 8% by NMR (Table 3). A smaller number have been investigated by other techniques, such as time-resolved crystallography.
Inferred Motions
Thus far, the discussion has focused only on "well-documented" motions, where high-resolution structures of at least two conformations (i.e. open and closed) are known. However, there is also the situation where one knows a single conformation of a given protein (A) is similar in structure to another protein (B) and that protein B has a well-documented motion. In this case, one can reasonably infer that protein A has a similar motion to that in protein B. Inferred motions are principally added to the database by finding sequence or structure homologues of a protein or nucleic acid already in the database. The inference is currently expressed at the top level in the preliminary classification scheme (Figure 2). For instance, heat-shock protein 70 is classified as having a "suspected shear motion" because of its structural similarity to hexokinase, which has a well-documented shear motion (31, 66). Furthermore, the motions initially suspected in actin and phosphoglycerate kinase based on analogy to other proteins (i.e. hexokinase) have been subsequently verified by crystallography (12, 22, 39, 43).
Motions can also be inferred based on a single known conformation and evidence based on requirements for the macromolecule’s function, careful calculations, or small-angle scattering experiments. Examples include the motions in myosin (84), plasminogen (70), and acetylcholinesterase (41). In total, about 78% of the motions have solved structures available for two or more conformations; for the remaining 22% the motions are inferred.
Computer Implementation as a Relational Database
Standard tools and approaches are currently used in the implementation of the database. A free relational database server engine, called mini-SQL (52), has been used with a schema that contains ~20 tables. Data entry has been done through a variety of methods: a web form, Microsoft Access and Excel (using ODBC connectivity or the dbf2msql program), or via the emacs text editor (101) (using a custom "mode" written in elisp). Initially, the web pages were generated "on the fly" in response to a query but then it was decided to pre-build most of them. This proved to be an unexpectedly good move as it allowed on-line search engines to automatically build indices up (e.g. AltaVista), enabling the database to be easily queried from outside. Because it is built using very standard tools, the database has been easily ported into a variety of programs (e.g. Oracle) and into a variety of PC mail-merge programs (for nicely formatted output). Although we plan to maintain pre-built pages in the future, we are investigating the use of high-speed web-database connectivity software (such as Informix’s Web datablade) to allow instantaneous updates to the database’s Web presence yet maintain a level of performance comparable to static pages.
In total, the database presently contains many disparate types of information: standardized annotation values, literature references, large blocks of free-text, three-dimensional structures, and motion pathways. This presents a particular challenge in terms of integrating the information in a comprehensible format. At present, many of the elements (e.g. movies) are stored outside of the central database (and accessed via stored pointers) or in the actual tables as large binary objects ("BLOBS"). We are presently migrating the database to an object-relational system made by Informix, a commercial product that traces its roots to the postgres database project at Berkeley (60, 90, 105). The object-relational database model supports the referencing of complex data types in relational tables and sophisticated querying of these complex types through user-defined functions. There are also plans to develop a data dictionary for the database around mmCIF (13).
Representing Motion Pathways as "Morph Movies"
One of the most interesting of the complex data types kept in the database are "morph movies" giving a plausible representation for the pathway of the motion. These movies can immediately give the viewer an idea of whether the motion is a rigid-body displacement or involves significant internal deformations (as in tomato bushy stunt virus versus citrate synthase). Pathway movies were pioneered by Vonrhein et al. (109), who used them to connect the many solved conformations of adenylate kinase.
Normal molecular-dynamics simulations (without special techniques, such as high temperature simulation or Brownian dynamics (55, 71, 112)) cannot currently approach the time scales of most of the motions in the database, which are estimated to be from several nanoseconds (loop closure) to several seconds (slow refolding) (26, 71, 74). Consequently a pathway movie cannot be generated directly via molecular simulation alone. Rather, it is constructed as an interpolation between known endpoints (usually two crystal structures). The interpolation can be done in a number of ways.
(i) Straight Cartesian interpolation. The difference in each atomic coordinate (between the known endpoint structures) is simply divided into a number of evenly spaced steps, and intermediate structures are generated for each step. This was the method used by Vorhein et al. It is easy to do, only requiring that the beginning and ending structures be intelligently positioned by fitting on a motionless core (34). However, it produces intermediates with clearly distorted geometry.
(ii) Interpolation with restraints. This is the above method where each intermediate structure is restrained to have correct stereochemistry and/or valid packing. One simple approach is to energy minimize each intermediate (with only selected energy terms) using a molecular mechanics program, such X-PLOR (16). This technique will be described more fully in a forthcoming paper (Krebs & Gerstein, in preparation). The database, furthermore, is currently home to an experimental server that applies this interpolation technique to two arbitrary structures, generating a movie.
Conclusion and Future Directions
We have constructed a database of macromolecular motions, which currently documents >120 motions. To describe each motion we have developed a classification scheme based on size then packing (whether or not there is motion across a well-packed interface) and a standardized nomenclature, such as maximum atomic displacement or degrees of rotation. We have also developed a way of annotating and categorizing inferred motions.
At present, many of the standardized statistics are culled from the literature, and most of the classification is done by eye. However, in the future much of the annotation will be done automatically with software tools. In particular, we are developing tools to objectively determine standardized statistics for a motion, produce "morph movies," locate flexible linkers using amino-acid composition or crystallographic temperature factors, classify motions, and cross-reference new motions to manually annotated "gold-standards" (using sequence and structure comparison).
We anticipate that the database will constitute an important resource for the molecular biology community. In fact, we expect that the number of macromolecular motions will greatly increase in the future, making a database of motions increasingly valuable. The reasoning behind this conjecture is as follows: The number of new structures continues to go up at a rapid rate (nearly exponential). However, the increase in the number of folds is much slower and is expected to level off much more in the future as the we find more and more of the limited number of folds in nature, estimated to be as low as 1000 (15, 23). Each new structure solved that has the same fold as one in the database represents a potential new motion -- i.e. it is often a structure in a different liganded state or a structurally perturbed homologue. Thus, as we find more and more of the finite number of folds, crystallography and NMR will increasingly provide information about the variability and mobility of a given fold, rather than identify new folding patterns.
Acknowledgements
The authors gratefully acknowledge the financial support of the National Science Foundation (Grant DBI-9723182) and the numerous people who have either contributed entries or information to the database or have given us feedback on what the user community wants. The authors also wish to thank Informix Software, Inc. for providing a grant of its database software.
References
1. No Author. (1997). Obstacles of nomenclature [editorial] [see comments]. Nature 389, 1.
2. Abad-Zapatero, C, Griffith, J P, Sussman, J L & Rossman, M G (1987). Refined Crystal Structure of Dogfish M4 Apo-lactate Dehydrogenase. J. Mol. Biol. 198, 445-467.
3. Abola, E, Sussman, J, Prilusky, J & Manning, N (1997). Protein Data Bank archives of three-dimensional macromolecular structures. Meth. Enz. 277, 556-571.
4. Altman, R B, Abernethy, N F & Chen, R O (1997). Standardized representations of the literature: combining diverse sources of ribosomal data. Ismb 5, 15-24.
5. Anderson, B F, Baker, H M, Norris, G E, Rumball, S V & Baker, E N (1990). Apolactoferrin structure demonstrates ligand-induced conformational change in transferrins. Nature 344, 784-787.
6. Anderson, C M, Zucker, F H & Steitz, T (1979). Space-filling models of kinase clefts and conformation changes. Science 204, 375-380.
7. Bairoch, A & Boeckmann, B (1992). The Swiss-Prot Protein-Sequence Data-Bank. Nucl. Acids Res. 20, 2019-2022.
8. Bairoch, A, Bucher, P & Hofmann, K (1996). The prosite database, its status in 1995. Nucleic Acids Research 24, 189-196.
9. Bennett, W S & Huber, R (1984). Structural and Functional Aspects of Domain Motion in Proteins. Crit. Rev. Biochem 15, 291-384.
10. Bennett, W S, Jr & Steitz, T A (1978). Glucose induced conformational change in yeast hexokinase. Proc. Natl. Acad. Sci. U.S.A. 75, 4848-4852.
11. Berman, H M, et al. (1992). The nucleic acid database a comprehensive relational database of three-dimensional structures of nucleic acids. Biophys J 63, 751-759.
12. Blake, C C F, Rice, D W & Cohen, F E (1986). A "helix-scissors" mechanism for the hinge-bending conformational change in phosphoglycerate kinase. Int. J. Peptide Protein Res. 27, 443-448.
13. Bourne, P E, Berman, H M, McMahon, B, Watenpaugh, K D, Westbrook, J & Fitzgerald, P M D (1997). The Macromolecular Crystallographic Information File (mmCIF). Meth. Enz. 277, 571-590.
14. Boutonnet, N S, Rooman, M J & Wodak, S J (1995). Automatic analysis of protein conformational changes by multiple linkage clustering. J. Mol. Biol. 253,
15. Brenner, S E, Chothia, C & Hubbard, T J (1997). Population statistics of protein structures: lessons from structural classifications [In Process Citation]. Curr Opin Struct Biol 7, 369-376.
16. Brünger, A T (1993). X-PLOR 3.1, A System for X-ray Crystallography and NMR (Yale University Press, New Haven).
17. Bullough, P A, Hughson, F M, Skehel, J J & Wiley, D C (1994). Structure of influenza haemagglutinin at the pH of membrane fusion [see comments]. Nature 371, 37-43.
18. Cate, J H, et al. (1996). Crystal structure of a group I ribozyme domain: principles of RNA packing [see comments]. Science 273, 1678-1685.
19. Chan, D C, Fass, D, Berger, J M & Kim, P S (1997). Core structure of gp41 from the HIV envelope glycoprotein. Cell 89, 263-273.
20. Chasman, D I, Flaherty, K M, Sharp, P A & Kornberg, R D (1993). Crystal Structure of Yeast TATA-Binding Protein and Model for Interaction with DNA. Proc. Natl. Acad. Sci. 90, 8174-8178.
21. Chen, R O, Felciano, R & Altman, R B (1997). RIBOWEB: linking structural computations to a knowledge base of published experimental data. Ismb 5, 84-87.
22. Chik, J K, Lindberg, U & Schutt, C E (1996). The structure of an open state of beta-actin at 2.65 A resolution. J Mol Biol 263, 607-623.
23. Chothia, C (1992). Proteins — 1000 families for the molecular biologist. Nature 357, 543-544.
24. Chothia, C, Lesk, A M, Dodson, G G & Hodgkin, D C (1983). Transmission of conformational change in insulin. Nature 302, 500-505.
25. Chuprina, V P, Rullmann, J A, Lamerichs, R M, J. H. van Boom, Boelens, R & Kaptein, R (1993). Structure of the complex of lac repressor headpiece and an 11 base-pair half-operator determined by nuclear magnetic resonance spectroscopy and restrained molecular dynamics. J Mol Biol 234, 446-462.
26. Creighton, T E (1984). Proteins (Freeman, San Francisco).
27. Donne, D G, et al. (1997). Structure of the recombinant full-length hamster prion protein PrP(29–231): The N terminus is highly flexible. Proc. Natl. Acad. Sci. USA 94, 13452–13457.
28. Epstein, J A, Kans, J A & Schuler, G D (1994). WWW Entrez: A Hypertext Retrieval Tool for Molecular Biology. 2nd Ann. Int. WWW Conf. (in press).
29. Evans, P R (1991). Structural aspects of allostery. Curr. Opin. Struc. Biol. 1, 773-779.
30. Fermi, G & Perutz, M F (1981). Haemoglobin and Myoglobin (Claredon Press, Oxford).
31. Flaherty, K M, McKay, D B, Kabsch, W & Holmes, K C (1991). Similarity of the three-dimensional structures of actin and the ATPase fragment of a 70-kDa heat shock cognate protein. Proc. Natl. Acad. Sci. USA 88, 5041-5045.
32. Genick, U K, et al. (1997). Structure of a protein photocycle intermediate by millisecond time- resolved crystallography. Science 275, 1471-1475.
33. Gerstein, M (1997). A Structural Census of Genomes: Comparing Bacterial, Eukaryotic, and Archaeal Genomes in terms of Protein Structure. J. Mol. Biol. 274, 562.
34. Gerstein, M & Altman, R (1995). Average core structures and variability measures for protein families: Application to the immunoglobulins. J. Mol. Biol. 251, 161-175.
35. Gerstein, M & Chothia, C (1996). Packing at the Protein-Water Interface. Proc. Natl. Acad. Sci. USA 93, 10167-10172.
36. Gerstein, M & Chothia, C H (1991). Analysis of Protein Loop Closure: Two Types of Hinges Produce One Motion in Lactate Dehydrogenase. J. Mol. Biol. 220, 133-149.
37. Gerstein, M & Hegyi, H (1998). Comparing Microbial Genomes in terms of Protein Structure: Surveys of a Finite Parts List. FEMS Microbiology Reviews (in press).
38. Gerstein, M, Lesk, A M, Baker, E N, Anderson, B, Norris, G & Chothia, C (1993). Domain Closure in Lactoferrin: Two Hinges produce a See-saw Motion between Alternative Close-Packed Interfaces. J. Mol. Biol. 234, 357-372.
39. Gerstein, M, Lesk, A M & Chothia, C (1994). Structural Mechanisms for Domain Movements. Biochemistry 33, 6739-6749.
40. Gerstein, M, Schulz, G & Chothia, C (1993). Domain Closure in Adenylate Kinase: Joints on Either Side of Two Helices Close Like Neighboring Fingers. J. Mol. Biol. 229, 494-501.
41. Gilson, M K, et al. (1994). Open "Back Door" in a Molecular Dynamics Simulation of Acetylcholinesterase. Science 263, 1276-1278.
42. Gregoret, L M & Cohen, F E (1990). Novel method for the rapid evaluation of packing in protein structures. J Mol Biol 211, 959-974.
43. Harlos, K, Vas, M & Blake, C F (1992). Crystal Structure of the Binary Complex of Pig Muscle Phosphoglycerate Kinase and Its Substrate 3-Phospho-D-Glycerate. Proteins: Struc. Func. Genet. 12, 133-144.
44. Harpaz, Y, Gerstein, M & Chothia, C (1994). Volume Changes on Protein Folding. Structure 2, 641-649.
45. Harrison, P M, Bamborough, P, Daggett, V, Prusiner, S B & Cohen, F E (1997). The prion folding problem. Curr Opin Struct Biol 7, 53-59.
46. Hogue, C W, Ohkawa, H & Bryant, S H (1996). A dynamic look at structures: WWW-Entrez and the Molecular Modeling Database. Trends Biochem Sci 21, 226-229.
47. Holm, L & Sander, C (1994). The FSSP database of structurally aligned protein fold families. Nuc. Acid Res. 22, 3600-3609.
48. Holm, L & Sander, C (1996). Mapping the Protein Universe. Science 273, 595-602.
49. Hubbard, S J & Argos, P (1994). Cavities and packing at protein interfaces. Protein Science 3, 2194-2206.
50. Hubbard, S J & Argos, P (1996). A functional role for protein cavities in domain-domain motions. J. Mol. Biol. 261, 289-300.
51. Hubbard, T J P, Murzin, A G, Brenner, S E & Chothia, C (1997). SCOP: a structural classification of proteins database. Nucleic Acids Res 25, 236-239.
52. Hughes, D (1996). mini-SQL program. http://Hughes.com.au.
53. Janin, J & Wodak, S (1983). Structural domains in proteins and their role in the dynamics of protein function. Prog. Biophys. Mol. Biol. 42, 21-78.
54. Johnson, L N & Barford, D (1990). Glycogen Phosphorylase. J. Biol. Chem. 265, 2409-2412.
55. Joseph, D, Petsko, G A & Karplus, M (1990). Anatomy of Conformational Change: Hinged ‘Lid’ Motion of the Triosephosphosphate Isomerase Loop. Science 249, 1425-1428.
56. Kim, Y, Geiger, J H, Hahn, S & Sigler, P B (1993). Crystal structure of a yeast TBP/TATA-box complex. Nature 365, 512-520.
57. Knowles, J R (1991). Enzyme catalysis: not different, just better. Nature 350, 121-124.
58. Knowles, J R (1991). To build an enzyme... Phil. Trans. R. Soc. Lond. B 332, 115-121.
59. Koonin, E V, Tatusov, R L & Rudd, K E (1996). Protein Sequence Comparison at a Genome Scale. Meth. Enz. 266, 295-322.
60. Korth, H & Silberschatz, A (1991). Database system concepts, 2nd edition (McGraw-Hill, New York).
61. Koshland, D E, Jr (1958). Proc. Natl. Acad. Sci. USA 44, 98-104.
62. Koshland, D E (1973). Protein Shape and Biological Control. Sci. Am. 229, 52-64.
63. Kraulis, P J (1991). MOLSCRIPT - A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24, 946-950.
64. Kuntz, I D (1992). Structure-Based Strategies for Drug Design and Discovery. Science 257, 1078-1082.
65. Lawson, C L, Zhang, R, Schevitz, R W, Otwinowski, Z, Joachimiak, A & Sigler, P B (1988). Flexibility of the DNA-Binding Domains of the trp Repressor. Proteins 3, 18-31.
66. Lesk, A M & Chothia, C (1984). Mechanisms of Domain Closure in Proteins. J. Mol. Biol. 174, 175-191.
67. Lesk, A M & Chothia, C (1988). Elbow Motion in the immunoglobulins involves a molecular ball and socket joint. Nature 335, 188-190.
68. Levitt, M, Gerstein, M, Huang, E, Subbiah, S & Tsai, J (1997). Protein Folding: the Endgame. Ann. Rev. Biochem. 66, 549-579.
69. Lewis, M, et al. (1996). Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science 271, 1247-1254.
70. Mangel, W F, Lin, B & Ramakrishnan, V (1990). Characterization of an extremely large, ligand-induced conformational change in plasminogen. Science (Washington D C) 248, 69-73.
71. McCammon, J A & Harvey, S C (1987). Dynamics of Proteins and Nucleic Acids (Cambridge UP,
72. McPhalen, C A, Vincent, M G, Picot, D, Jansonius, J N, Lesk, A M & Chothia, C (1992). Domain closure in mitochondrial aspartate aminotransferase. J. Mol. Biol. 227, 197-213.
73. Meador, W E, Means, A R & Quiocho, F A (1992). Target enzyme recognition by Calmodulin: 2.4 Å structure of a Calmodulin-Peptide Complex. Science 257, 1251-1255.
74. Moffat, K (1989). Time-resolved macromolecular crystallography. Annu Rev Biophys Biophys Chem 18, 309-332.
75. Murzin, A, Brenner, S E, Hubbard, T & Chothia, C (1995). SCOP: A Structural Classification of Proteins for the Investigation of Sequences and Structures. J. Mol. Biol. 247, 536-540.
76. Newman, M, Strzelecka, T, Dorner, L F, Schildkraut, I & Aggarwal, A K (1995). Structure of Bam HI endonuclease bound to DNA: partial folding and unfolding on DNA binding. Science (Washington D C) 269, 656-663.
77. Olson, A J, Bricogne, G & Harrison, S C (1983). Structure of Tomato Bushy Stunt Virus: The Virus Particle at 2.9 Å Resolution. J. Mol. Biol. 171, 61.
78. Orengo, C A, Jones, D T & Thornton, J M (1994). Protein superfamilies and domain superfolds. Nature 372, 631-634.
79. Peretz, D, et al. (1997). A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol 273, 614-622.
80. Perutz, M (1989). Mechanisms of cooperativity and allosteric regulation in proteins. Quart. Rev. Biophys. 22, 139-236.
81. Pley, H W, Flaherty, K M & McKay, D B (1994). Three-dimensional structure of a hammerhead ribozyme [see comments]. Nature 372, 68-74.
82. Ponder, J W & Richards, F M (1987). Tertiary templates for proteins: use of packing criteria in the enumeration of allowed sequences for different structural classes. J. Mol. Biol. 193, 775-791.
83. Povey, S, White, J, Nahmias, J & Wain, H (1997). Problems of nomenclature [letter; comment] [see comments]. Nature 390, 329.
84. Rayment, I, et al. (1993). Three-dimensional Structure of Myosin Subfragment-1: A Molecular Motor. Science 261, 50-58.
85. Rees, B, Cavarelli, J & Moras, D (1996). Conformational flexibility of tRNA: structural changes in yeast tRNA(Asp) upon binding to aspartyl-tRNA synthetase. Biochimie 78, 624-631.
86. Remington, S, Wiegand, G & Huber, R (1982). Crystallographic refinement and atomic models of two different forms of citrate synthase at 2.7 and 1.7 Å resolution. J. Mol. Biol. 158, 111-152.
87. Richards, F M (1977). Areas, Volumes, Packing, and Protein Structure. Ann. Rev. Biophys. Bioeng. 6, 151-176.
88. Richards, F M (1985). Calculation of Molecular Volumes and Areas for Structures ofKnown Geometry. Methods in Enzymology 115, 440-464.
89. Richards, F M & Lim, W A (1994). An analysis of packing in the protein folding problem. Quart. Rev. Biophys. 26, 423-498.
90. Rowe, L A & Stonebraker, M R (1987). The POSTGRES data model, in Proceedings of the Thirteenth International Conference on Very Large Data Bases: 1987 13th VLDB. (eds. Stocker, P M, Kent, W & Hammersley, P) 83-96 (Morgan Kaufmann, Los Altos, CA, USA).
91. Ruff, M, et al. (1991). Class II aminoacyl transfer RNA synthetases: crystal structure of yeast aspartyl-tRNA synthetase complexed with tRNA(Asp). Science 252, 1682-1689.
92. Sampson, N S & Knowles, J R (1992a). Segmental Movement: Definition of the Structural Requirements for Loop Closure in Catalysis by Triosphosphate Isomerase. Biochemistry 31, 8482-8487.
93. Sayle, R & Milner-White, E J (1995). RasMol: Biomolecular graphics for all. Trends in Biochemical Sciences 20, 374.
94. Schlichting, I, et al. (1990). Time-resolved X-ray crystallographic study of the conformational change in Ha-Ras p21 protein on GTP hydrolysis. Nature 345, 309.
95. Schmidt, R, Gerstein, M & Altman, R (1997). LPFC: An Internet Library of Protein Family Core Structures. Prot. Sci. 6, 246-248.
96. Schuler, G D, Epstein, J A, Ohkawa, H & Kans, J A (1996). Entrez: Molecular Biology Database and Retrievel System. Meth. Enz. 266, 141-162.
97. Scott, W G, Finch, J T & Klug, A (1995). The crystal structure of an all-RNA hammerhead ribozyme: a proposed mechanism for RNA catalytic cleavage. Cell 81, 991-1002.
98. Shilton, B, Flocco, M, Nilsson, M & Mowbray, S (1996). Conformational changes of three periplasmic receptors for bacterial chemotaxis and transport: the maltose-,glucose/galatactose- and ribose-binding proteins. J. Mol. Biol. 264, 350-363.
99. Shon, K J, Kim, Y, Colnago, L A & Opella, S J (1991). NMR studies of the structure and dynamics of membrane-bound bacteriophage Pf1 coat protein. Science 252, 1303-1305.
100. Silicon-Graphics (1996). VRML 2 Specification. http://webspace.sgi.com/moving-worlds/Design.html.
101. Stallman, R (1986). GNU Emacs Manual (Free Software Foundation Inc., Cambridge, MA).
102. Stein, P & Chothia, C (1991). Serpin tertiary structure transformation. J. Mol. Biol. 221, 615-621.
103. Stevens, R C, Gouaux, J E & Lipscomb, W N (1990). Structural consequences of effector binding to the T state of aspartate carbamoyltransferase: crystal structures of the unligated and ATP- and CTP- complexed enzymes at 2.6 A resolution. Biochemistry 29, 7691-7701.
104. Stevens, R C & Lipscomb, W N (1992). A molecular mechanism for pyrimidine and purine nucleotide control of aspartate transcarbamoylase. Proc. Natl. Acad. Sci. 89, 5281-5285.
105. Stonebraker, M R & Rowe, L A (1986). The Design of POSTGRES, in Proc. 1986 ACM-ACM-SIGMOD Conf. on Management of Data Int. Conf. on Mgt. of Data.
106. Stryer, L (1995). Biochemistry (W H Freeman and Company, New York).
107. Subramaniam, S, Gerstein, M, Oesterhelt, D & Henderson, R H (1993). Electron diffraction analysis of structural changes in the photocycle of bacteriorhodopsin. EMBO J. 12, 1-8.
108. Tsai, C J, Lin, S L, Wolfson, H J & Nussinov, R (1996). A dataset of protein-protein interfaces generated with a sequence-order-independent comparison technique. Journal Of Molecular Biology 260, 604-620.
109. Vonrhein, C, Schlauderer, G J & Schulz, G E (1995). Movie of the structural changes during a catalytic cycle of nucleoside monophosphate kinases. Structure 3, 483-490.
110. Vyas, N K, Vyas, M N & Quiocho, F A (1991). Comparison of the periplasmic receptors for L-arabinose, D-glucose, and D-ribose — structural and functional similarity. J. Biol. Chem. 266, 5226-5237.
111. Wade, N (1997). Scientists Find A Key Weapon Used by H.I.V. New York Times, Saturday, April 19, 1997, A1 and 9.
112. Wade, R C, Davis, M E, Luty, B A, Madura, J D & McCammon, J A (1993). Gating of the active site of triose phosphate isomerase: Brownian dynamics simulations of flexible peptide loops in the enzyme. Biophys. J. 64, 9-15.
113. Wierenga, R K, Noble, M E M, Postma, J P M, Groendijk, H, Kalk, K H, Hol, W G J & Opperdoes, F R (1991). The crystal structure of the "open" and the "closed" conformation of the flexible loop of trypanosomal triosephosphate isomerase. Proteins 10, 93.
114. Williams, N (1997). How to get databases talking the same language [news]. Science 275, 301-302.
Table 1 Standard Statistics for the Magnitude of the Motions
|
Value |
Num. Entries |
min |
max |
average |
|
Maximum C a displacement |
11 |
1.5 |
60 |
12 |
|
Maximum Atomic Displacement |
3 |
8.8 |
10 |
9.3 |
|
Maximum Rotation |
12 |
5 |
148 |
24 |
|
Maximum Translation |
2 |
0.7 |
2.7 |
1.7 |
The motions in the database range greatly in size, with maximum mainchain displacements between 1.5 and 60 Å. All the statistics are for version 1.7 of the database, based on the relatively small set of values culled from the literature. The averages are only approximate given the sparse nature of the data. We are developing software tools to extract these values automatically from structural data.
Table 2 Statistics for the Mechanism of the Motions
This table cross tabulates the two main classifying attributes of motions: their size (row heads) and their packing characteristics (column heads). We define a known motion (**) to be a motion with two or more solved conformations, and a suspected motion is defined to have only one or fewer solved conformations.
Table 3 Statistics for the Evidence about Motions
|
Experimental Technique |
Entries studied by this technique |
Fraction of database |
|
All Techniques |
122 |
100% |
|
Traditional X-ray crystallography |
116 |
95% |
|
NMR |
9 |
7% |
|
Molecular Dynamics Simulations |
4 |
3% |
|
Time-resolved crystallography |
3 |
2% |
|
Circular Dichroism (CD) |
2 |
2% |
|
Fourier Transform Infrared Spectroscopy (FTIR) |
1 |
<1% |
|
Molecular Biology Studies of Motion |
1 |
<1% |
This table summarizes the number of motions studied by the various experimental techniques. We indicate the evidence behind a motion through listing information about the experimental techniques used, telling whether or not the motion is inferred, and giving a standardized "annotation level." We also timestamp all entries with creation and modification dates and associate the web presentation of the database with a clear version numbering scheme. Note percentages in this table do not add up to 100% as a motion can be studied by more than one technique.
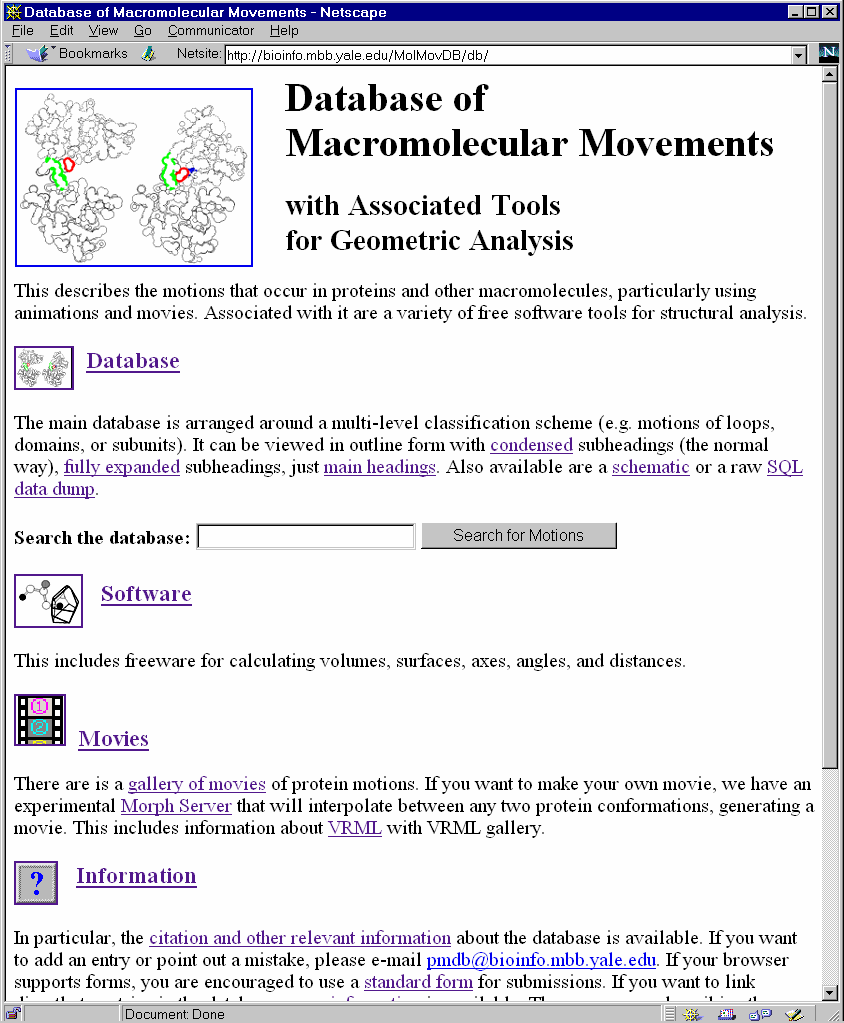
Fig. 1 The Motions Database on the Web
LEFT shows the World Wide Web "home page" of the database. One can type keywords into the small box at the top to retrieve entries. RIGHT shows an entry retrieved by such a keyword search (the entry for calmodulin). Graphics and movies are accessed by clicking on an entry page. (These have been deliberately segregated from the textual parts of the database since the interface was designed to make it easy to use on a low-bandwidth, text-only browser, e.g. lynx or the original www_3.0). An example of a segregated graphic for calmodulin is the movie shown in Figure 5. The main URL for the database is http://bioinfo.mbb.yale.edu/MolMovDB. Beneath this are pages listing all the current movies, graphics illustrating the use of VRML to represent endpoints, and an automated submission form to add entries to the database. The database has direct links to the PDB for current entries (http://www.pdb.bnl.gov); the obsolete database for out-of-date entries (http://pdbobs.sdsc.edu); scop for structure classification (http://scop.mrc-lmb.cam.ac.uk); Entrez/PubMed for literature citations (http://www.ncbi.nlm.nih.gov/PubMed); LPFC for core structures, (Library of Protein Family Core Structures, http://smi-web.stanford.edu/projects/helix/LPFC); and GeneCensus for information related to structural genomics (http://bioinfo.mbb.yale.edu/census) (3, 75, 95, 96). Through these links one can easily connect to other common protein databases such Swiss-Prot, Pro-Site, CATH, RiboWeb, and FSSP (4, 7, 8, 21, 47, 78). For all these links, PDB identifiers or PubMed unique IDs are used as foreign keys. External databases may also link to entries in the motions database by using PDB identifiers as foreign keys. In particular, the interface to the database is via the following URL convention: http://bioinfo.mbb.yale.edu/MolMovDB/search.cgi?pdb=1abc, where 1abc is a PDB structure identifier referenced in the movements database. Further, information on the database's public interface and on linking external resources to it may be obtained by at http://bioinfo.mbb.yale.edu/MolMovDB/linkhelp.txt . We are developing transaction-processing features that allow authorized remote experts to serve as database editors and anticipate that these will become an important part of the interface in the future. (This figure is as well as Figures 2, 3, 4, and 5 are adapted directly from the web presentation of the database, which is copyright, Gerstein & Krebs, 1998).
Fig. 2 Schematic Showing the Overall Classification Scheme for Motions
LEFT, the database is organized around a hierarchical classification scheme, based on size (fragment, domain, subunit) and then packing (hinge or shear). Currently, the hierarchy also contains a third level for whether or not the motion is inferred. RIGHT is a schematic showing the difference between shear (sliding) and hinge motions. This figure adapted from the database and refs. (38, 39). It is important to realize that the hinge-shear classification in the database is only "predominate" so that a motion classified as shear can contain a newly formed interface and one classified as hinge can have a preserved interface across which there is motion. The essential characteristics of the various motions are summarized below. To annotate a macromolecule's classification succinctly a three-letter short-hand code is used. It designates the major classification (Fragment, Domain, Subunit, Complex, or Nucleic acid), sub-classification (hinge, shear, allosteric, non-allosteric, RNA, or DNA), and whether or not the motion has been solved structurally in at least two conformations. For example, ‘D-h-2’ would indicate a domain hinge motion with at least two conformations solved.
|
Shear Mechanism |
Hinged Mechanism |
|
|
Well-Packed Interfaces |
MAINTAINED, throughout motion |
NOT MAINTAINED; Rather created, burying surface |
|
Mainchain Packing |
Constrained by close packing |
Free to kink at hinge |
|
Mainchain Torsions |
Many small changes |
A few large changes |
|
Motion Overall |
Concatenation of small local motions |
Identical to twisting at hinge |
|
Motion at Interface |
Parallel to plane of interface (shear) |
Perpendicular to interface |
|
Sidechain Packing |
Same packing in both forms |
New contacts; |
|
Sidechain Torsions |
Mostly small changes |
Some large changes |
|
Simple Example |
Trp Repressor, Insulin |
Lactoferrin, Calmodulin |
Fig. 3 Close-up on the Shear Mechanism
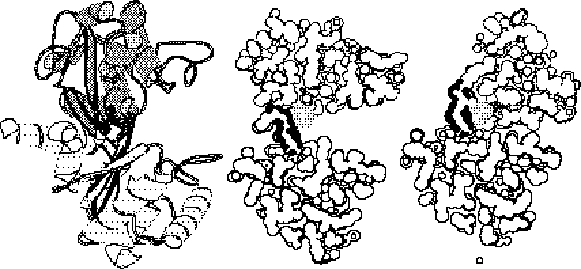
The figure gives a close up illustrating shear motion in one protein, citrate synthase (39, 66). TOP-LEFT and TOP-RIGHT show representative shear motions between close-packed helices. Note how the mainchain only shifts by a small amount and the sidechains stay in the same rotamer configuration. MIDDLE-LEFT, Cartoon of one subunit of citrate synthase (1CTS), gives an overall view of the protein showing that it is composed of many helices. The adjacent subunit is related by two-fold axis shown. (The small two-stranded sheet is omitted to improve clarity.) a-helices are represented by cylinders. The small domain contains helices N, O, P, Q, and R. The mobile OP helix is highlighted. MIDDLE-RIGHT gives details on the mobile interfaces. The orientation is perpendicular to the twofold axis. The particular section is indicated by the dotted line on the MIDDLE-LEFT subfigure. Selected helixes from both subunits are shown. (Upper-case letters are for one subunit and lower-case letters are for the other one.) The helices shown with white lettering on a black background are motionless, while those shown in black on white move appreciably. Edges indicate the existence of helix-helix packing in both the open and closed form. Double edges are nearly parallel packing (0-30°); single edges, intermediate packing (30°-60°); and dotted edges, crossed packing (60°-90° and on-end packing). There is no packing between helixes L and N because helixes L, M, G, and F are much higher (coming out of page) than O, N, Q, P, R, and K. S and I are long and make contacts with both sets. Note in the diagram how the dimer neatly divides into six layers with the active site, indicated by a star, at the intersection between layers. This is representative of how proteins undergoing shear motions can be divided into layers. Part of one subunit is enlarged at the bottom of the diagram and shows the relative movements of the principal helices in citrate synthase. The shifts (in Angstroms) and rotations (in degrees) show local changes in the positions of pairs of packed helices (i.e. the movement in one helix in a pair relative to the other). Clearly, larger relative movements tend to be associated with more crossed helix-helix packing. BOTTOM shows how these small motions can be added together to produce a large overall motion. Specifically, many small motions add up to shift helix O by 10.1 Å and rotate it by 28°. The incremental motion in shear domain closure is shown by Ca traces of the whole protein and of a close-up of the OP loop. BLACK is the apo form; WHITE, holo form; GRAY, cumulative effect of motion over the K, P, and then Q helix-helix interfaces. (The apo form was fit to the holo form, first on the core, and then on the K, P, and Q helices.)
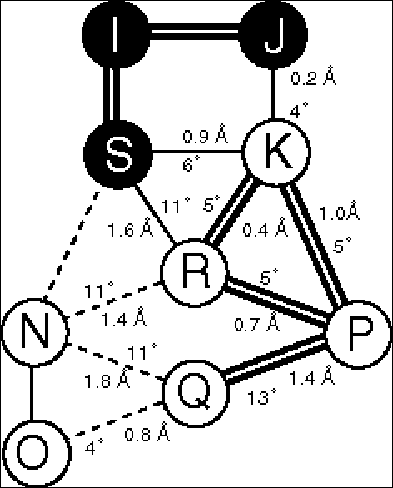
Fig. 4 Close-up on the Hinge Mechanism
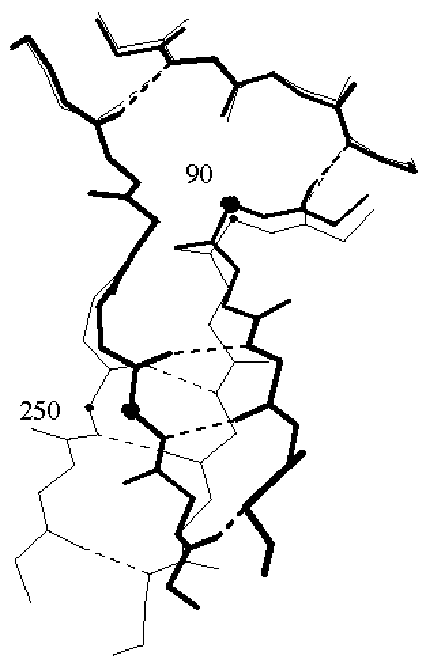
The figure shows the hinge motion in lactoferrin (38, 39). FAR-LEFT shows a ribbon drawing of the protein in the open conformation. The view is down the screw-axis, which is indicated in the figure by the circle with the dot in it. The screw-axis passes very close to the hinge region, which occurs in the middle of two beta strands (highlighted in bold). MIDDLE-LEFT and MIDDLE-RIGHT show the open and closed conformations in terms of space filling slices. A thick black line highlights the hinge region. Note how few packing constraints there are on the hinge in contrast to the other atoms in the protein. FAR-RIGHT shows a close-up of the hinge region. (The numbered residues correspond to the open circles in the ribbon drawing.) (Figure adapted from the database and ref. (38)).
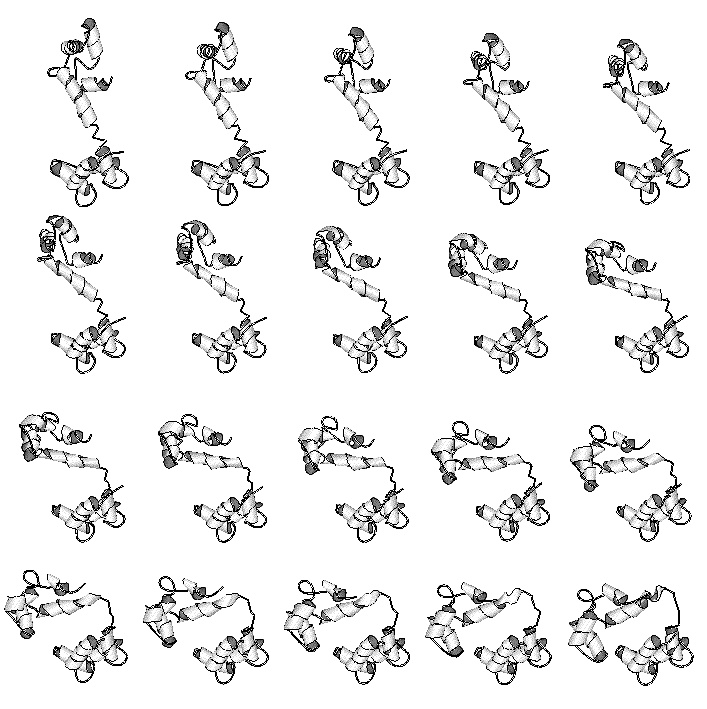
Fig. 5 Interpolated Motion Pathways
A preliminary pathway of the hinge motion in the protein calmodulin is shown (73). This was constructed by a variant of the second method, involving Cartesian interpolation with minimization of the intermediate structures using both stereochemical and packing terms. This and more than 30 other movies are available at http://bioinfo.mbb.yale.edu/MolMovDB/movie . For the actual generation of representations, currently one orientation is chosen (i.e. down the screw-axis) and then the animated intermediates are drawn in a variety of 2D-movie formats (MPEG, QuickTime, SGI movie format, MultiGIF, and so on). Preliminary 3D animation has been implemented using the new VRML-2 specification (100); however, we have encountered some compatibility problems due to the great state of flux that VRML 2.0 browser software presently is in.
Calmodulin, which is shown in Figure 1 as well as in this figure, is one of the more highly annotated motions in the database. It provides a good example of how the overall annotation process works. A motion is initially brought to our attention either directly by researchers solving particular structures or indirectly by surveying the literature. Once we decide to add it to the database, we do a comprehensive literature search, usually via Medline, and retrieve from the original publications statistics associated with the motion. It is in itself quite a complex nomenclature problem to reconcile the many different terms used to describe motion and create truly standardized statistics (such as a well-defined maximum atomic displacement or precise selections for hinge residues). This is one aspect of the larger problem of nomenclature that is becoming increasing important in bioinformatics (1, 83). Next, we fetch coordinate sets from the PDB and run various comparison programs on these structures (e.g. to calculate torsion angle differences, do least-squares fits, evaluate packing, etc.). Part of the process of conformation comparison is the generation of a "morph movie," such as the one shown in the figure. Our server (Krebs & Gerstein, in preparation) can produce a morph completely automatically. Typically, two structures are selected as being representative of the endpoints of the motion. Intermediate conformations are generated from these endpoints by linear interpolation with restraints applied at each interpolated time point to ensure realism. (For the case of calmodulin, bond length and angle restraints were applied.) The interpolated coordinates are joined into an animation through using any of a number of widespread molecular rendering software packages (e.g. Molscript or Rasmol (63, 93)). Morphing and automatic conformation comparison generates a second, more standardized set of statistics, which can be compared against those culled from the literature. Finally, based on running programs and reading the literature, we decide on the motion classification and write the entry. Presently, much of this process is done manually but we hope to automate large amounts of it in the future. The automatic classification tool developed by Boutonnet et al. (14) may be useful in this regard. Because our database schema is flexible, it can readily accommodate different types of automatic and manual annotation.
Fig. 1 The Motions Database on the Web
|
|
|
|
Fig. 2 Schematic Showing the Overall Classification Scheme for Motions
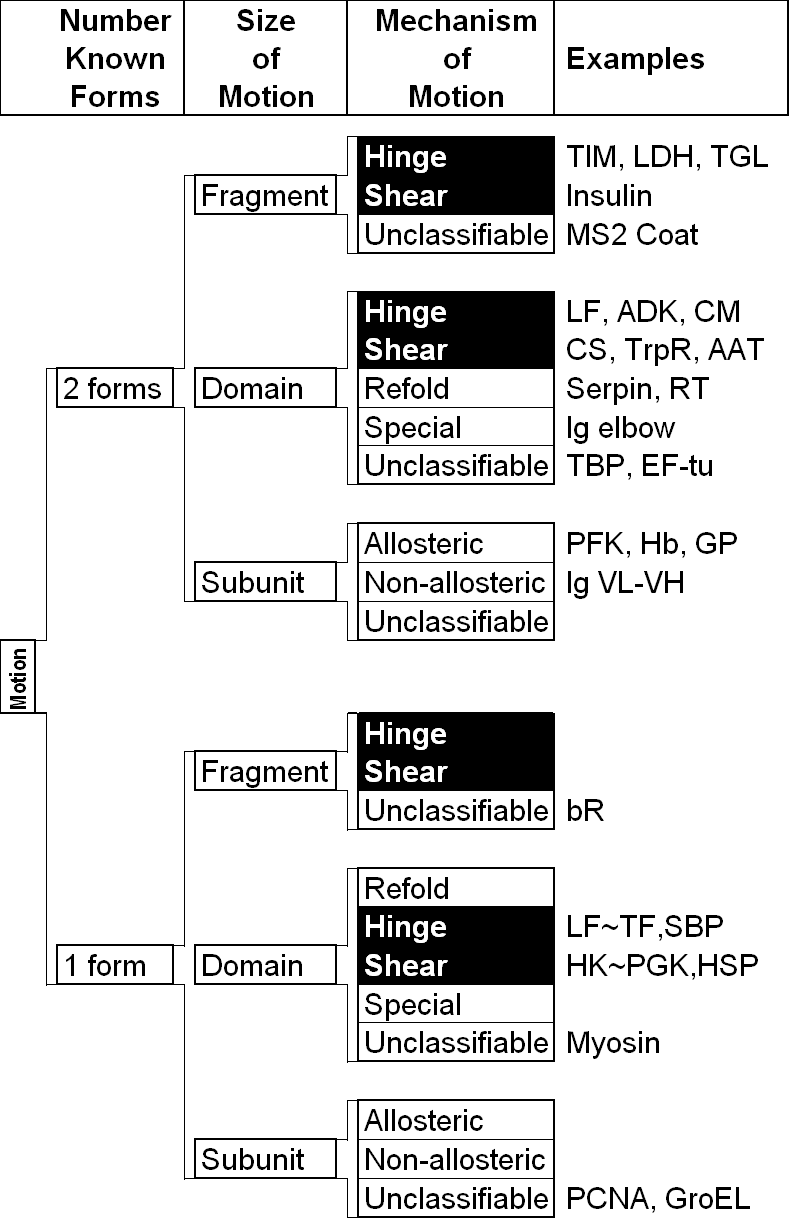
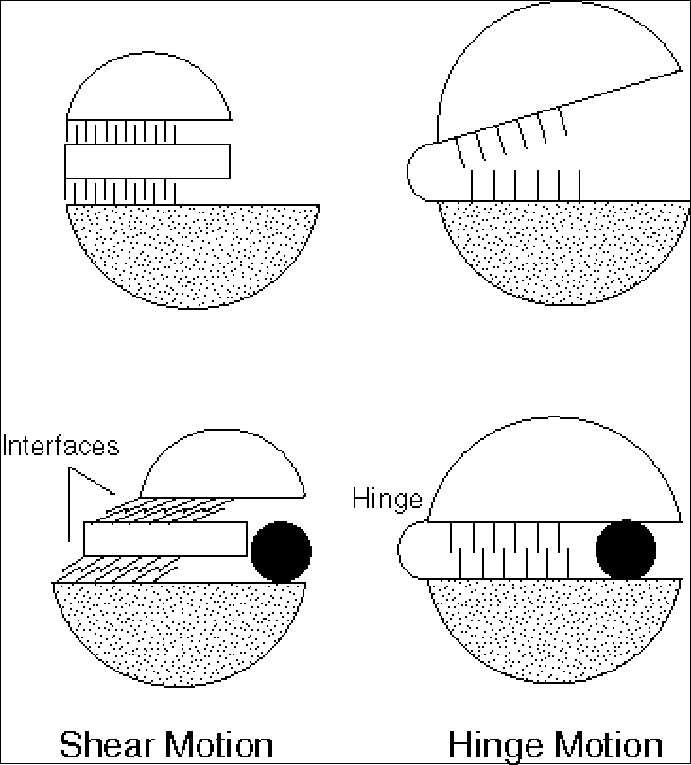
Fig. 3 Closeup on the Shear Mechanism
|
|
|
|
|
|
|
|
|
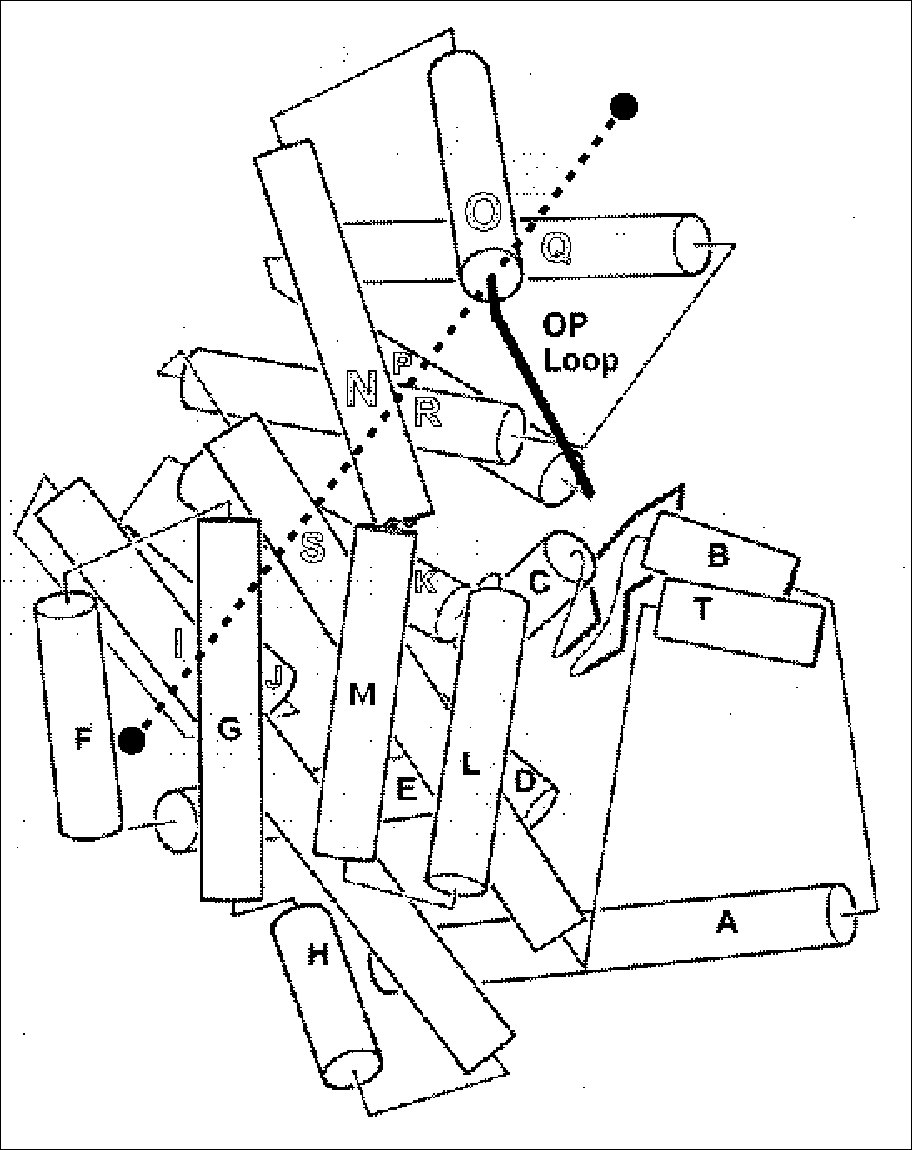
Fig. 4 Closeup on the Hinge Mechanism
|
 |
|
|
Fig. 5 Interpolated Motion Pathways