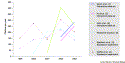| Register or Login: | Password: | |||||||
|
|
|
|
|||||||||||||||||||||||||||||||||||
| ||||||||||||||
| This Document | ||
| SummaryPlus | ||
| Full Text + Links | ||
| PDF (63 K) | ||
| Actions | ||
| Cited By | ||
| Save as Citation Alert | ||
| E-mail Article | ||
| Export Citation | ||
Editorial overview
Sequences and topology
Mark Gerstein
Janet M Thornton
Molecular Biophysics and Biochemistry Department, Bass 432A, 266 Whitney Avenue, Yale University, New Haven, CT 06520, USA
European Bioinformatics Institute, Wellcome Trust Genome Campus, Cambridge CB10 1SD, UK
Available online 11 June 2003.
![]()
 Introduction Global genome analysis Family-level genome analysis Structural genomics and fold assignments Functional genomics analysis (expression) Functional genomics analysis (genome-scale protein–protein interaction networks) Protein–protein interactions and three-dimensional docking Finding sites in three-dimensional structures Finding sites in three-dimensional structures I An overall focus on protein–protein interaction networks II References
Introduction Global genome analysis Family-level genome analysis Structural genomics and fold assignments Functional genomics analysis (expression) Functional genomics analysis (genome-scale protein–protein interaction networks) Protein–protein interactions and three-dimensional docking Finding sites in three-dimensional structures Finding sites in three-dimensional structures I An overall focus on protein–protein interaction networks II ReferencesIn what follows, we give an overview of the eight reviews in the Sequences and topology section, describing and summarizing their content. Basically, the reviews this year follow the overall progression in bioinformatics from genome sequence to functional genomics analysis to protein structure. We start with reviews by Karlin and Lancet on genome analysis, the former focusing on more global issues and the latter on what can be learned from specific families. Then, the review by Orengo discusses mapping from the genome to protein families and structure. Stolovitzky talks about analyzing expression information, functional genomics data that characterize the sequence products. The next group of reviews, by Eisenberg and Janin, discuss protein–protein interactions, the interactome, and the current excitement in interpreting this. Finally, we have reviews by Jackson and Westhead, and Nakamura on more detailed aspects of three-dimensional site analysis.
Karlin, Mrázek and Gentles survey three classes of amino acid features revealed through the recent genome sequencing efforts. Reviewed are occurrence frequencies of several sequence features with potential evolutionary and structural import. Findings are put in the context of the evolutionary tree of life. As demonstrated, doing so can provide evidence for or against standing evolution-related theories.
Sequence homology can provide insight into a gene product's structure. Lancet and co-workers focus on this using the olfactory receptors as a case study. For instance, within gene families, the most variable amino acids in their multiple alignments can be considered as potential active sites. These sites are likely to have mutated to acquire related but different functions. Interspecies homologies also have shed light on structure, as it is likely that functional residues will be conserved. Other comparative studies attempt to identify the role single nucleotide polymorphisms may play structurally, possibly finding clues to their role in complex genetic diseases.
The continuous stream of completed genome sequences offers interesting insights into protein evolution. As the number of completed genomes increases, it is becoming clear that the plethora of proteins occurring in nature are built from a relatively few conserved structural elements. Work in this field has been able to identify ancient evolutionary linkages not necessarily apparent from considering sequence data alone. Orengo and co-workers review discoveries along these lines, as well as the computational approaches (and their limitations) used to obtain them.
Microarray technology enables us to assess the differential expression between two mRNA samples of tens of thousands of genes in parallel. Along with this vast amount of data comes the sizeable task of assessing the levels of differential expression in a rigorous way. A rich literature has arisen to deal with this question and is reviewed by Stolovitzky. It is suggested that the resulting lists of discriminating genes can vary from method to method. Because of this, it is important for researchers to understand the methods available, as different algorithms are naturally better suited to different problems.
The study of protein–protein interactions is reviewed by Salwinski and Eisenberg as a tight interplay between experimentation and bioinformatics. The role of computational methods covers a priori interaction predictions, interactions validation and the analysis of resulting interaction networks.
Janin and Séraphin review recent technological advances that suggest the majority of proteins exist and function as part of large protein complexes. This discovery necessitates new experimental and bioinformatics approaches for evaluating protein–protein interactions. Whereas traditional approaches towards elucidating protein–protein docking mainly deal with binary interactions, newly developed approaches must attempt to determine the assembly of multicomponent complexes. Several computational methods to this end are noted, as is the importance of assessing their reliability. Reviewed are the efforts of the community-wide Critical Assessment of Predicted Interactions (CAPRI) experiment, which provides a venue for evaluating docking prediction schemes.
Jackson, Westhead and co-workers review computational approaches towards understanding protein–ligand binding. First, they discuss approaches to identifying protein active sites. Approaches discussed are those utilizing the growing abundance of protein family sequence and structural data in evolution-derived prediction schemes, as well as methods employing biophysical and biochemical calculations, such as electrostatics, pK shifts and thermodynamics. Techniques for assessing functional site similarity are also reviewed. Methods considered involve structural template matching and clique detection algorithms. Finally, advances in ligand docking, docking scoring functions and docking validation are addressed.
Important tasks for protein informatics are delineated by Kinoshita and Nakamura. Recent advances in functional site identification via evolutionary means, and biophysical and biochemical calculations are first reviewed. Next, methods to predict the biochemical function of uncharacterized proteins are offered. These predictions can be achieved by comparing newly identified or predicted active sites with those of known function, or by focusing on properties of molecular surfaces. Lastly, techniques for gleaning a protein's overall biological role are summarized.
Overall, one of the striking features about this year's reviews was their focus on protein–protein interaction networks, in particular, the networks developed through the recent in vivo pull-down experiments by Gavin et al. [1] and Ho et al. [2]. Networks clearly have the attention of those in the computation of biology.
This is borne out, to some degree, by looking at Figure 1, in which we plot the number of citations of three different types of major data sets: expression data sets (dotted lines), structure data sets (dashed lines) and protein–protein interaction data sets (solid lines). The expression sets are represented by the classic papers by Spellman et al. [3] on the yeast cell cycle and DeRisi et al. [4] on the diauxic shift. Structure data are represented by the ribosome structure [5], clearly a capstone achievement in macromolecular crystallography. Interaction data were represented initially by the Uetz et al. two-hybrid paper [6], and now by the Gavin et al. [1] and Ho et al. [2] in vivo pull-down papers.
Figure 1. Citations to major data sets of relevance to bioinformatics.
One can clearly see how the structure and expression citations, to
some degree, are leveling off and there is a tremendous jump in
citations for the interactions data sets. These citation data reflect a
shift in biology as a whole, from molecules ultimately to complete
systems. The new data present very exciting challenges for
computational biology, to shape hypotheses and interpret the living
world.
![]()
1. A.C. Gavin, M. Bosche, R. Krause, P. Grandi, M. Marzioch, A. Bauer, J. Schultz, J.M. Rick, A.M. Michon, C.M. Cruciat et al., Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415 (2002), pp. 141–147. Abstract-Elsevier BIOBASE | Abstract-EMBASE | Abstract-GEOBASE | Abstract-MEDLINE | Full Text via CrossRef
2. Y. Ho, A. Gruhler, A. Heilbut, G.D. Bader, L. Moore, S.L. Adams, A. Millar, P. Taylor, K. Bennett, K. Boutilier et al., Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415 (2002), pp. 180–183. Abstract-BIOTECHNOBASE | Abstract-Elsevier BIOBASE | Abstract-EMBASE | Abstract-MEDLINE | Abstract-GEOBASE | Full Text via CrossRef
3. P.T. Spellman, G. Sherlock, M.Q. Zhang, V.R. Iyer, K. Anders, M.B. Eisen, P.O. Brown, D. Botstein and B. Futcher, Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 12 (1998), pp. 3273–3297. Abstract-BIOTECHNOBASE | Abstract-EMBASE | Abstract-MEDLINE
4. J.L. DeRisi, V.R. Iyer and P.O. Brown, Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278 (1997), pp. 680–686. Abstract-MEDLINE | Abstract-EMBASE | Abstract-BIOTECHNOBASE | Full Text via CrossRef
5. N. Ban, P. Nissen, J. Hansen, P.B. Moore and T.A. Steitz, The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289 (2000), pp. 905–920. Abstract-BIOTECHNOBASE | Abstract-Elsevier BIOBASE | Abstract-INSPEC | Abstract-EMBASE | Abstract-MEDLINE | Full Text via CrossRef
6. P. Uetz, L. Giot, G. Cagney, T.A. Mansfield, R.S. Judson, J.R. Knight, D. Lockshon, V. Narayan, M. Srinivasan, P. Pochart et al., A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403 (2000), pp. 623–627. Abstract-EMBASE | Abstract-MEDLINE | Abstract-BIOTECHNOBASE | Abstract-Elsevier BIOBASE
|
|
||||||||||||||||||||||||||||||||||
Volume 13, Issue 3 , June 2003 , Pages 341-343 |
||||||||||||||||||||||||||||||||||
|
Send feedback to ScienceDirect
|
 |