Editorial Overview for
2001 Current Opinion in Structural Biology
Issue on
Sequences and Topology
Mark Gerstein and Barry Honig
The articles
appearing in this issue of COSB deal with many of the data sources currently used
in bioinformatics – genome sequences, three dimensional structures of proteins,
and expression datasets. They also include a wide variety of computational approaches - sequence and structure
alignment and analysis, gene-expression clustering, and biophysical analysis.
Broadly, the issue follows the molecular-biological "data flow" from
raw genome sequence to detailed structural understanding, in the process
touching on genome annotation, integration of expression information, fold
assignments, structural alignment, and the understanding of protein-protein
interactions.
Bioinformatics is a new field and is still in
the process of being defined. It is focused on analysis of genomic, and more
recently, proteomic data. The range of tools that has been brought to bear on
these data is enormous and, as reflected in the articles in this issue, have
their sources in different disciplines; computer science, mathematics and
statistics, physics, chemistry, biochemistry and biophysics. A remarkable
aspect of the growth of bioinformatics has been the speed in which the various
disciplines have been integrated into the research programs of individual labs.
For example, biophysicists have learned and even developed dynamic programming
methods while computer scientists have made important contributions to the
analysis of three dimensional structure. A common language is emerging, some
common goals exist while others are in the process of being defined.
As is necessarily the cases, different
perspectives are evident in the articles in this issue and in the literature
that is reviewed. A common thread that runs through many of the articles is the
use of clustering to define biological “parts” and then the use of these parts
as frameworks for data integration and analysis. The clustering and
classification of data is of course an essential element in the history of
biology and the vast quantity of new data that has become available opens up a
seemingly limitless set of possibilities for the derivation of new relationships
and groupings. An alternate viewpoint emphasizes continuous aspects of
biological data. We will return to the apparent “conflict” between these two
perspectives below. A second issue that
arises from the emphasis on clustering involves the actual goals of the various
analyses being carried out. On the one hand the clustering of data and the
derivation of new relationships is a valuable goal in its own right. On the
other hand, it is not yet clear how to maximize the impact of bioinformatics on
mainstream biology which for many years has focused on the detailed
characterization of individual systems in specific organisms. It is worth
reading the articles in this issue with these questions and perspectives in
mind.
Califano discusses recent trends in sequence
analysis and describes how profile-based methods have in most cases replaced
pairwise analysis methods. Profile methods of course rely on the existence of
multiple alignments or on the ability to generate them on the fly, and reflect
major advances that have taken place in building consensus models for sequence
families. A second theme discussed in
this article is the combination and integration of different methods towards
various goals. Examples include the identification of regulatory motifs in
eukaryotes, protein structure prediction and the combined use of expression
clustering and sequence data in promoter detection.
The review by Kriventseva et al. addresses
the clustering and analysis of protein families in greater detail. They discuss
the construction and use of protein family databases and how a number of these
are integrated in the new Interpro resource. The article also discusses
clustering in structural databases and phylogenetic classification as well.
Kriventseva et al. emphasizes the challenge associated with the prediction of
protein function and highlight the gene ontology consortium whose goal is to
produce a vocabulary for biological processes.
It will be interesting to see the extent to which modern biology, and
biologists, will be amenable to the construction of a controlled vocabulary.
Altman & Raychaudhuri discuss various
applications of whole genome expression analysis. A variety of clustering
methods have been applied to microaray data and others have been developed.
They describe how the clusters found from analyzing expression data can be used
as a starting point for the prediction of regulatory elements, protein
function, interactions, and localization. In fact, the Altman &
Raychaudhuri's review artfully illustrates yet another application of
clustering, that of clustering literature databases. They base their entire
exposition on a clustering of expression analysis literature in terms of word
counts.
The review by Gaasterland & Opera
discusses a fundamental problem in genomics, how to identify proteins in raw
genome sequence. They provide a thoughtful discussion of the current state of
the annotation of some of the larger eukaryotes, in particular, human and fly.
They also discuss how the experimental evidence for verifying proposed
new genes -- e.g. ESTs, cDNAs,
microarrays, and homology matches -- integrates a number
different data sources.
Koehl discusses various methods that have
been developed to compare proteins in structural terms and highlights the
difficulties in arriving at a quantitative measure of protein structure
similarity. As discussed below, this is area where alternate viewpoints exist
in that structural classifications into discrete groupings are widely used
while there appears to be a continuous aspect to structural space as well.
Structural alignments can in many cases produce sequence alignments that are
superior to those obtained from pure sequence methods and there is clearly much
work to be done in integrating the two approaches, particularly as the amount
of three dimensional structural information continues to grow.
Teichmann et al. focus on the information to
be gained from large-scale structure determination, specifically structural
genomics projects on model organisms. The article offers a number of
interesting examples of the fact that structure reveals a wealth of functional
information and evolutionary relationships not available from sequence
alone. The article also focuses on the
use of different methods to deduce functional information. These range from the
physical properties of active sites to the use of phylogeny to predict
protein-protein interactions. Teichmann et al describe the assortment of
methods that are being used to predict what proteins interact with one another,
a problem that cannot be solved directly from structural genomics projects that
tend to focus on individual domains. An important lesson emphasized in the
article involves the limitations, at least at present, of automatic methods.
For example, the SCOP database for example, relies on human decision about
evolutionary relationships.
Protein-protein and protein-small molecule
interactions are the focus of the article by Ma et al. that also discusses how
structure comparison can be used to study protein flexibility and plasticity.
This article highlights the fact that fold of the polypeptide chain is only one
way of finding relationships between proteins. The nature of the protein
surface, where interactions actually occur, is another. Far less attention has
been paid to surface properties than to chain fold, but the situation is
changing fairly rapidly.
Membrane proteins are in some ways
fundamentally different than water proteins since they exist in part in a
non-polar environment. Ubarretxena-Belandia & Engelman discuss some of the
unique features of a-helical membrane
proteins from this perspective, emphasizing for example the relative abundance
of p-helical turns and the importance of
inter-helical hydrogen bonds. a-helical
membrane proteins illustrate how it is possible to generate considerable
functional diversity in a single structural framework.
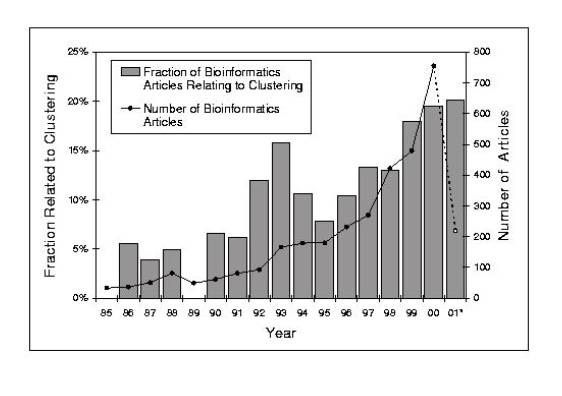
As mentioned above, many of the articles in
this issue emphasize clustering although rationale and mathematical basis for
the clustering process is often very different. One
can get a broader sense of the importance of clustering the databases into
parts than just indicated by the articles in this review by directly analyzing
the entirety of the recent bioinformatics literature, as catalogued by NCBI's
PubMed resource. The figure shows how the prevalence of papers related to
clustering and defining biological parts has increased markedly in the last few
years (since 1996).
Of course, clustering and grouping data is a general
mathematical issue confronted in a wide range of disciplines from psychology to
astronomy and many generally applicable techniques such as principal components
analysis have been developed. Some of the bioinformatics work, particularly
that in expression analysis, makes explicit reference to previously developed
approaches in computer science and statistics. However, in other areas one sees
more of a "home-brew" effort, often reflecting the special structure
of biological data. For instance, many of the methods in sequence clustering
are preoccupied with dealing the "multidomain problem," the way that
a protein with domain A could be inadvertently clustered with domain B through
multidomain protein AB.
In analyzing molecular biological information
in terms of clusters one confronts two very different perspectives. On one
hand, there may be unique clusters suggested by the data, with clear divisions
between each group. These, in turn, form natural foci for organizing databases
into discrete parts and integrating information. On the other hand, clustering
may be able to find groups in the data but these may not be unique. That is,
the division between two clusters may not involve crossing sharp boundaries but
rather might involve a continuous gradation. This suggests that partlist
concept maybe of only limited utility.
The alternative viewpoints have emerged most
clearly in the grouping protein structures into folds . There are number of
well-established fold classifications, such as CATH, SCOP, and FSSP, that
divide protein structures into a very limited catalogue of essential shapes
[1-3]. This "partslist" has enabled it to serve as a basis for the
integration of rich amount of heterogeneous biological information (see, in
particular, partslist.org [4]) and has encouraged speculation that in total
there is only a very small number of folds (e.g. ~1000-5000). However, a number
of recent papers have argued that "fold-space" is, in fact,
continuous and that a description in terms of unique, discrete parts may not be
possible [5,6]. It may be that both perspectives are valid and that they can be
used either together, or individually, in different applications. An analogy
that Janet Thornton has suggested involves the electromagnetic spectrum which
is clearly continuous but which can usefully divided for many purposes into
discrete wavelength regions with somewhat nebulous boundaries between
them.
A somewhat related issue arises in the
definition of function. Classification schemes exist here as well and these
will be expanded and integrated through efforts such as those of the gene
ontology consortium. Nevertheless, there are limits to functional
classification that are set in part by the limits of biological understanding.
Classification can clearly help in the discovery of relationships between
proteins and these are clearly extremely useful and important. On the other
hand, there are unique aspects to each protein, or pathway, that will resist
classification; for example binding specificity has a discrete element by its
very nature. Although the focus of much current bioinformatics research
involves classification, it may be equally useful to develop tools that make it
possible to bring a broad range of data and approaches to bear so as to focus
on the unique aspects of a particular problem, perhaps in the process allowing,
in a dynamic sense, for the discovery of new classifications and relationships.