Patterns of Protein-Fold Usage
in Eight Microbial Genomes:
A Comprehensive Structural Census
Mark Gerstein
Department of Molecular Biophysics & Biochemistry
266 Whitney Avenue, Yale University
PO Box 208114, New Haven, CT 06520
(203) 432-6105, FAX (203) 432-5175
Mark.Gerstein@yale.edu
Running Title: Comparing Genomes in terms of Protein Folds
Keywords: Structure Databank, Superfold, Protein Structure
Manuscript Length is: 28 Pages (including this one)
Graphics of Figures follow at end in sequence.
Submitted to: Proteins
Version: gb805
Abstract
Eight microbial genomes are compared in terms of protein structure. Specifically, yeast, H. influenzae, M. genitalium, M. jannaschii, Synechocystis, M. pneumoniae, H. pylori, and E. coli are compared in terms of patterns of fold usage -- whether a given fold occurs in a particular organism. Of the ~340 soluble protein folds currently in the structure databank (PDB), 240 occur in at least one of the eight genomes, and 30 are shared amongst all eight. The shared folds are depleted in all-helical structure and enriched in mixed helix-sheet structure compared to the folds in the PDB. The top-10 most common of the shared 30 are enriched in superfolds, uniting many non-homologous sequence families, and are also especially similar in overall architecture -- eight having helices packed onto a central sheet. In yeast the top-10 most highly duplicated folds are among the most highly expressed folds. However, they are very different from the common folds in the PDB, highlighting databank biases. Each of the eight organisms, even the minimal M. genitalium, has at least one unique fold not present in the others. A tree can be constructed grouping the genomes in terms of their shared folds. This is remarkably similar to more conventional classifications, based on very different measures of relatedness. Finally, folds of membrane proteins are analyzed through transmembrane-helix (TM) prediction. All the genomes appear to have similar usage patterns for these folds, with the occurrence of a particular fold falling off rapidly with increasing number of TM-elements, according to a "Zipf-like" law. This implies there are no marked preferences for proteins with particular numbers of TM-helices (e.g. 7-TM). Further information pertinent to this analysis is available at http://bioinfo.mbb.yale.edu/genome.
Introduction
In the last three years the genomes of a number of free-living organisms have been completely sequenced, generating tremendous interest, popular as well as scientific [1-3]. This event provides a unique opportunity to perform comprehensive comparisons between organisms on a molecular level. One of the most interesting questions that can be addressed through such comparisons is whether different organisms have distinctly different patterns of protein fold usage. That is, to what degree is there a common set of molecular parts (or shapes) that are shared universally amongst different organisms? Or, conversely, to what degree do certain protein folds occur only in one group of organisms and not in others (e.g. in eukaryotes but not in eubacteria)?
This type of "occurrence" analysis has been performed previously in terms of sequence motifs, families, functions, and biochemical pathways. Starting from the most basic units, genomes have been compared in terms of the relative frequencies of short oligonucleotide and oligopeptide "words" [4-7]. The degree of gene duplication in a number of genomes has been ascertained [8-13]. Other analyses have looked at how many highly conserved sequence families in one organism are present in another [14-19]. Finally, if sequences can be related to specific functions and pathways, one can see whether homologous sequences in two organisms truly have the same role (ortholog vs. paralog) and whether particular pathways are present or absent in different organisms [8, 16, 20-23]. This work has yielded many interesting conclusions in terms of pathways that are modified or absent in certain organisms. For instance, the essential citric acid cycle is found to be highly modified in H. influenzae [23, 24]. Furthermore, identifying pathways and proteins unique to certain microbes may prove useful for developing drugs (e.g. antibiotics against bacteria, [16]). In some genome annotation systems, attempts have been made to integrate a variety of analyses and perform them on a large scale in a highly automated fashion [18, 25-29].
The analysis of structure and fold families is expected to be particularly advantageous from the point of view of occurrence analyses for three reasons:
First, structures allow one to more precisely define the "module" or part that is shared. This is particularly true for groups of aligned structures, which allow the definition of a structural core [30, 31]. It is possible (and quite productive) to define modules purely in terms of conserved regions in sequence alignments [32-38]. However, functioning protein modules fundamentally consist of units of 3D structure, usually folding domains, and relating modules defined on the sequence level to structure enables them to be better characterized.
Secondly and more importantly, one expects analysis of structure to reveal more about distant evolutionary relationships than just sequence comparison, since structure is more conserved than sequence or function [39, 40]. In other words, it is at the level of protein structure where one sees the greatest redundancy and reuse in biology. It is believed that the number of structural motifs is very limited, and elucidation of this limited repertoire of molecular parts is seen as one of the principal future challenges for biology [41, 42].
A final reason that structure is advantageous for genome comparisons is that the relationship between sequence similarity and structural similarity is much better defined than the corresponding relationship between sequence and function.
It is generally accepted that proteins with similar sequences usually have similar structures. A decade ago Chothia & Lesk systematically investigated this relationship [39, 43]. They found that the extent of the structural changes is directly related to the extent of the sequence changes. The relationship between sequence similarity and functional similarity is much less clear [44]. In part, this is because it is much more difficult to precisely specify a function than a sequence or a structure. Moreover, even in cases where the functional identification is well specified, there are several examples where highly similar sequences have completely different functions - i.e. same fold but different function.
Here the eight genomes listed in Table 1 are compared in terms of their usage of protein folds. These eight, which are amongst the first to be completely sequenced, provide a most diverse comparison: They represent microbes from the three kingdoms of life (Eukaraya, Eubacteria, Archaea), from different environments (room temperature and pressure to high temperature and pressure, and neutral pH to highly acidic), with a wide range of genome sizes (0.6 to 13 Mb), and with a variety of modes of life (from parasite to autotroph).
The comparisons here follow up on recent work comparing fold usage in representative collections of sequences from different species or in complete inventories of predicted structures in a genome [12, 45, 46]. There has also been much work focussing specifically on surveying the occurrence of membrane proteins in genomes [12, 47-55]. As the work here implicitly involves comparison of protein structures, it also rests upon a foundation provided by the emerging protein fold classifications [56-63].
Categories of Folds
The protein folds in the genomes can be divided into three categories:
(1) Those Corresponding to Known Structures of Soluble Proteins. Based on current technology, folds in this category represent 6% to 14% of the total residues in the genomes, 9% on average (involving 11-20% of the ORFs). Similar fractions have been found in many previous analyses [28, 44]. These folds are the part of the genome that can be best characterized in terms of protein structure and will be dealt with first, in the next section.
(2) Those Made from Transmembrane Helices. Folds in this category are analyzed by transmembrane helix prediction.
(3) Other. Proteins in this category are either soluble proteins with (currently) unknown fold, membrane proteins composed of transmembrane beta-strands (such as porins [64]), or proteins that do not assume a fixed conformation (such as repetitive, low-complexity regions [65]). While some of these proteins can be surveyed structurally to a limited degree by prediction methods, here they are filtered out and excluded from the analysis. (For an example of the identification of beta-membrane proteins in genomes, see Champion et al. [66].)
Division of the PDB into Families, Folds and Superfolds
An important preliminary step in characterizing the known folds in the genomes is clustering the proteins in the structure databank (the PDB [67]) into sequence families, groups of homologous sequences for which there is no significant similarity between groups. Doing this, via a new clustering approach described in the methods, gives 990 distinct sequence families in the (current) databank. Then, using the structural similarity relationships in the scop database, sequence families that share the same fold but which have no detectable homology can be combined into folds. There are currently 338 folds in the PDB, with an average of three sequence families per fold [68]. The fact that the number of folds is considerably less than the number of sequence families suggests that many of the evolutionary similarities between highly diverged organisms may only be apparent in terms of structure, all the sequence similarity having been eroded away [69].
The known folds can be ranked by how many different families of non-homologous sequences they are associated with. Folds uniting many distinct sequence families have been dubbed superfolds [59]. These may represent intrinsically stable and favorable structural arrangements, as suggested by a variety of analyses [59, 70, 71]. Here the 25 known folds associated with the most sequence families are defined to be superfolds.
Thus the analysis begins by dividing the structure databank at three levels: into 990 sequence families, which are apportioned amongst 338 folds, which, in turn, contain 25 superfolds.
Analysis of Soluble-Protein Folds in Genomes
Fold Tables and Usage Patterns in terms of Binary Numbers
Having been clustered, the known structures in the PDB were compared against the eight genomes. The raw results take the form of two "fold" tables, listing how many times each of the 990 sequence families and 338 folds in the PDB occur in each of the eight genomes. The complete tables are quite large (8x338 and 8x990), so showing them in full is not possible. Only the top quarter of one is reproduced here, listing the 54 folds that occur in at least seven of the eight genomes (Figure 1). However, as described in the methods section, the complete tables (and other associated information) are available over the web in a variety of convenient formats (see, in particular, http://bioinfo.mbb.yale.edu/genome/browser/fold-report).
The raw fold tables are condensed and cross tabulated into summaries (Tables 2 to 3), indicating how often particular patterns of fold usage occur (i.e. how often a fold is in yeast and E. coli but not in the other six genomes). One way of achieving this condensation is through the use of Venn diagrams [45, 72, 73]. However, this is awkward for eight genomes. A more convenient representation for these patterns is through an 8-digit binary number, where a digit is "1" if the fold occurs in the corresponding genome and "0" if it does not (and "*" matches both occurs and does not). There are 255 possible patterns of fold usage (28-1). However, as indicated in Table 2, only about a quarter (62, 24%) of these patterns are observed.
The most common single pattern of fold usage is for a fold to occur in all eight genomes, and this occurs 30 times as shown in Figure 1. The 30 shared folds presumably represent a most ancient and essential set of molecular parts, as they are present in all three kingdoms of life, in a wide variety of environments, and in genomes of very different size. They include a number of ribosomal protein folds (e.g. L14 and S5, domain 2), folds that act as scaffolds for many different functions (TIM-barrel and OB-fold), folds for the binding of cofactors and certain other molecules (NAD-binding Rossmann fold and the thiamin-binding fold), and folds associated with specific metabolic functions (the phosphoglycerate kinase fold and the P-loop containing NTP hydrolase fold). There are fewer sequence families than folds present in all the genomes (26), dramatically illustrating how structure is conserved more than sequence.
Another common and simple to understand fold-usage pattern is when a fold is present in only a single genome, i.e. for folds unique to specific organisms. As shown in Table 3, each of the eight genomes has at least one unique fold. Yeast has the most unique folds followed by EC (23 then 19). On the other extreme, MJ has three unique folds, and MG, only one.
The converse of a fold being present in a single genome is for it to be absent from only one of the eight genomes. Twenty-four folds are present in exactly seven of the eight of the genomes. They are shown in Figure 1. It never occurs that a fold is missing from EC, SC, HI, or SS and is present in all the other genomes. When a fold is missing from one genome, it is usually missing from MJ (16 times out of 24). In a similar fashion, most of the time when a fold is present in six of the eight genomes it is absent from MP and MG.
Disregarding whether a fold occurs in another genome, one finds that 240 of the 338 known folds in the PDB (and 547 of the 990 sequence families) occur in at least one of the eight genomes. Thus, ~10% of the residues in these primitive organisms match ~70% of the known folds.
Overall, EC has the most distinct (not unique) folds, followed by SC, and predictably MG, MP, and MJ have the fewest. As a fraction of the number of its ORFs, MJ has the fewest known folds.
Fold Usage Tree
One can also take the observed patterns of fold usage and use this to cluster the genomes. A "distance" between two genomes can be reasonably defined as the number of common folds shared between two genomes as a fraction of the total folds in the genomes. This is similar to the definitions of distance used in traditional phylogenetic analysis, where the number of shared taxonometric characters or features is used as the basis for classification [74]. Other definitions are, of course, possible. A tree built with this distance metric is shown in Figure 3.
For comparison the fold-usage tree is shown next to a number of other trees constructed by different distance measures:
(1) the overall difference in amino acid over the whole genome;
(2) the number of shared sequence families (very similar to the number of shared folds) and;
(3) the sequence divergence of related proteins that share the same fold and are present in all 8 genomes. This last measure of distance is most similar to the customary measure based on individual proteins.
Remarkably, despite being derived from such different properties of the organisms, these trees are all very similar to each other in topology and similar to the conventional classification of microbes, based on 16S ribosomal RNA sequences (summarized in Table 1) [8, 75]. That is, they group together the gram-positive bacteria (MP and MG) and the gram-negative bacteria (HI and EC) and position these two bacterial lineages with the cyanobacteria SS a distance from the eukaryote SC and the archeon MJ. The major difference between the trees is the treatment of HP, which is closer to the mycoplasms in the composition tree and closer to EC and HI in the other trees. HP is a gram-negative proteobacteria and should be grouped with EC and HI. However, it has been found to be rather problematical in terms of evolutionary classification [53, 76].
Distribution of Fold Classes and Superfolds
To gain more structural insight into what types of folds are shared between genomes, it is possible to classify each fold as all-a, all-b, a/b, a+b, or other (using the definitions of Levitt & Chothia [77]) and then to see how the folds corresponding to each of the structural classes are distributed amongst the genomes (Figure 1 and Tables 3 and 4). As compared to the PDB, the most common folds in the genomes (i.e. those that occur in many genomes) are enriched in mixed domains containing both helices and strands and depleted in all-alpha ones. Specifically, of the 30 folds that occur in all eight genomes one is all-alpha, four are all-beta and the rest are mixed (3% all-alpha and 13% all-beta). In contrast, of the 338 folds in the PDB, 75 are all-alpha and 55 are all-beta (22% and 16%). EC has most of the unique all-beta folds, and yeast, the most unique all-alpha ones.
The superfolds are much more highly represented in the folds present in all (or many) genomes than they are in the PDB. In particular, seven of the 25 superfolds are present in all eight genomes, and only two are not present in at least one genome. Figure 2 shows how superfolds are shared to a greater degree between genomes than are folds, and likewise, how folds are shared more than sequence families.
Number of Structure Matches and the Top-10 Folds
In addition to being ordered by how many genomes they occur in, folds can also be arranged by how often they occur in total in all eight genomes. That is, the number of "matches" that PDB structures with a given fold have in all eight genomes can be used to rank the folds. The 240 folds present in at least one genome have 3610 total matches in all the genomes, about 15 per fold and 2 per fold per genome. The yeast genome contributes the most structure matches of the eight genomes (1073), reflecting its large size and highly duplicated character [11].
The ten folds of the thirty that occur most often in all eight genomes, i.e. those with the most matches, are drawn in Figure 4. These are the top-10 folds. They include the seven superfolds that are present in all eight genomes. Furthermore, they have a remarkably similar architecture, containing interleaved helices and sheets. They can be divided into barrel folds (reductase/elongation-factor, OB-fold, and TIM-barrel), classic alpha/beta folds with helices packed on either side of a central sheet (P-loop hydrolase, Rossmann fold, and Thiamin-binding), folds with helices packed onto a single face of a sheet (ferrodoxin, FAD-binding, and beta-grasp), and a fold with a more complex structure (class II synthetase). Overall, eight of the top-10 contain a clear central sheet with helices packed onto at least one face. The two exceptions are the OB-fold and the reductase/elongation-factor fold, which are mostly structured by strands.
Ranking Folds by Expression Level
The top-10 list in figure 4 ranks folds by how often they occur in the genome, tending to emphasize highly duplicated genes. Folds can also be ordered by a number of other criteria. In particular, they can be ranked in terms of expression level, essentially by mRNA occurrence in the cell. This has already been done in non-structural terms for all the genes in yeast [78-80]. Table 4 shows how this expression level ranking maps onto folds. Using data from DeRisi et al. [80], the table shows the most highly expressed folds in yeast grown in two different conditions (high sugar and low sugar, aerobic vs. anaerobic conditions). The ranking of folds is clearly different from that purely based on duplication.
Analysis of Membrane-Protein Folds in Genomes
Overall Numbers
The usage of membrane protein folds was surveyed by first performing simple transmembrane-helix (TM) predictions and then seeing what structural class the prediction was in, i.e. 3-TM, 5-TM, and so on.
Overall, about 5% of the residues in the genomes are in transmembrane helices, ranging from a high of 7% in EC to a low of 3% in MJ. The number of ORFs with at least one transmembrane element ranges from ~35% in EC, SC, and SS to ~20% in MJ for an average of 28%, indicating that EC, SC, and SS have more membrane proteins (as a fraction of the total) than the other genomes. This agrees with previous work by others, where it was found that ~20-30% of the proteins in microbial genomes are membrane proteins, the specific value depending somewhat on prediction method and threshold used [12, 47-55].
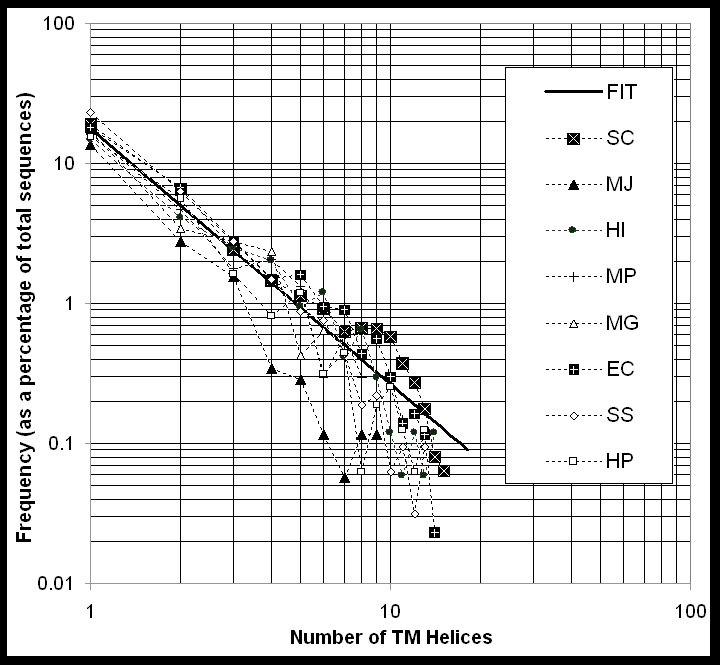
Zipf's Law Fit and 7-TM proteins
The number of TM-helices per protein follows a similar decreasing pattern in each genome, with fewer proteins having large numbers of TM-helices. As shown in Figure 5, the fraction F of proteins in the genome with a given number n of TM-helices can be fit with the expression F(n) = .18 n-1.8, where n ranges from 0 to 15. (Without great degradation of the fit, the even simpler expression 1/[5n2] can be used as well.) This expression has a form like that of the Zipf's Law that often occurs in the analysis of word frequency in documents [81]. Similar Zipf-law-like expressions have been found to apply in a variety of other situations relating to the occurrence of proteins (e.g. in relation to the occurrence of oligopeptide words [82-84]). Moreover, this particular functional form for the occurrence of proteins with a given number of TM-helices falls off smoothly with an increasing number of helices (n), implying that there is no particular preference (i.e. local maximum) for proteins with seven TM-helices. This suggests that this heavily studied group of proteins is not exceptionally important in the context of microbial genomes.
There are two additional points to note about Figure 5: First, the logarithmic scale of the figure tends to emphasize proteins with many TM-helices. However, this can be a bit deceptive as the bulk of the membrane proteins in each genome have only a few TM-spans (e.g. 2-TM). Second, while the frequency of TM-helices fits the Zipf's law fairly well in an overall sense, on closer examination there are some notable differences between the genomes. In particular, MP appears to have more 7-TM folds than average (3% v 1%) and EC more 12-TM folds. Yeast has fewer 9-TM helix folds, and MG, fewer 10- and 11-TM folds.
Most of the membrane-protein surveys agree on this absence of 7-TM proteins in microbial genomes; some also claim to find more 6 and 12 TM proteins in bacterial genomes corresponding to well known families of transporter proteins [12, 50, 52, 55]. In contrast, surveys of the incomplete (and highly biased) set of human sequences and the unfinished worm genome find a relative abundance of 7-TM proteins in these multicellular organisms [50, 55].
Conclusion
Summary
Eight microbial genomes have been compared in terms of their usage of protein folds. To this end, a "binary-number" representation was developed for counting and comparing patterns of fold usage. It was found that the eight genomes contain 240 of the 338 known soluble protein folds, and 30 of these are shared amongst all eight. As compared to the PDB, the shared folds are enriched in mixed structure (both helices and sheets) and depleted in all-alpha domains. The ten most common of the 30 shared folds (the top-10) are especially similar in structure, with eight of the ten having a classic architecture of helices packed against a central sheet. They are also particularly enriched in superfolds (7 of 10).
Each of the eight genomes, including the very minimal MG, has at least one unique protein fold not present in any of the others. Conversely, when a fold is absent from only one genome, it is usually absent from the archeon MJ. Overall, a tree clustering the genomes in terms of their number of shared folds has a remarkably similar structure to more conventional classifications that are based on amino-acid composition or ribosomal sequences.
Finally, the folds of membrane proteins were analyzed through TM-helix prediction. All the genomes appear to have similar patterns of usage for these folds. The occurrence of a particular membrane-protein fold falls off rapidly with more TM-helices, according to a Zipf's law, and there are no marked preferences for folds with particular numbers of TM-helices (such as 7-TM proteins).
Limitations of the Approach: The Small, Incomplete Number of Known Folds
There are a number of limitations to the fold-usage analysis presented here. First, only a relatively small number of folds can be surveyed, involving no more than a fifth of the ORFs in a genome. (This number would be even smaller if one were to restrict attention to just the ORFs in a genome that have been studied directly by crystallography or NMR. For example, it is currently 52 out of 6218 for yeast, as reported by Sacch3D [85].)
The situation is expected to improve in the future as new structures are determined, but it will be a while before all the folds in a genome are known -- especially considering that the increase in new folds is much slower than the increase in new structures [68]. An important corollary of this is that the absolute counts found in a given genome survey are (usually) an under-representation of the true numbers. Furthermore, they are contingent on the evolving contents of the databank. Thus, over time as more structures are added to the databank, one should expect such statistics as the most common folds and number of shared folds to change somewhat.
Comprehensive application of ab initio structure prediction and advanced sequence-comparison and fold recognition methods to complete genomes can overcome somewhat the limitations of only knowing a small number of folds [12, 46], allowing one to survey the complete inventory of proteins in an organism. However, in its present form, structure prediction is not a substitute for structure determination, especially in situations where the fold is completely new. Moreover (as discussed below) using state-of-the-art sequence comparison methods introduces a measure of variability and uncertainty into the results, as different methods will give different results at the margin.
The uncertainties in the analysis resulting from the small number of known folds are aptly illustrated in table 5. This shows the top-10 folds in yeast calculated two ways, as done here versus as done for the Sacch3D database [85]. The differences between the two lists, which are described in detail in the table caption, are easily understood and well illustrate how the exact ordering of the top-10 list depends on two factors: the sequence-structure comparison methods used and the contents of the current database of folds. Different comparison programs and fold databases will give different numbers.
Limitations of the Approach: Biases in PDB and in the Genomes
In addition to rendering the results here, in a sense, incomplete, the small number of known folds also means that the results may be influenced to some degree by the biases in the PDB. These biases are manifest in a number of ways.
First and most simply, there is a considerable disparity between how often a fold occurs in the genomes (i.e. how many total matches it has) and how often it occurs in the PDB (i.e. how many known structures have this fold). This is indicated in Figure 1 (and in the web presentation). One can immediately see how different the common folds are in the PDB versus in the genomes. This illustrates in a direct sense the biases in the PDB -- although these sort of biases are not expected to affect the results (which are principally concerned with "membership" rather than absolute counts).
Second and more subtly, the composition of the PDB is biased towards folds that occur in more heavily studied organisms such as EC and SC. These biases are probably reflected in some of the results -- specifically, in the finding that there are many more known folds and unique folds in the bacterium HI than in the archeon MJ, even though both of these organisms have genomes of approximately the same size.
Another subtle bias in the results here is in the selection of genomes. The eight organisms picked were the first with complete genomes to be sequenced, as has by necessity been done in all the other multi-genome comparisons to date (e.g. [16]). A more balanced comparison would perhaps have a more comparable amount of eukaryotes and archaea to bacteria.
Prospects
Improvements in the results presented here will have to wait for more data, more genomes sequenced and more structures determined. Despite these limitations, comparisons of genomes in terms of protein structure are certain to yield results about the fundamental differences between organisms on a molecular level. There are currently more than 10 microbial genomes completed and at least 35 more being worked on [86], so there will be many possibilities for comparison soon.
Sequence and Structure Analysis Techniques
A Relational Database of Genome Sequences and Structure Assignments
Translated genome sequences were taken from the web sites (Table 1). The genome data is constantly changing and is contingent on the current "state of the art" in gene finding. The data used in this paper reflects a particular snapshot of this ongoing process. For instance, the E. coli data file used was version M52, containing 4290 ORFs. This is a more recent version and contains a different number of ORFs than one referred to in the official publication (M49, containing 4288 ORFs [87]). For yeast there is some uncertainty regarding whether all of the ORFs in the web site file are really genes. In particular, 5888 of the 6218 ORFs are definitely believed to be genes, but there is some question about the remaining 330 [88]. Furthermore, quite a number of yeast sequences (initially) annotated to be ORFs are, in fact, transposons, which should properly be segregated from the rest of the proteome [89]. ), the distinct spike around 100 residues is almost undoubtedly an artifact caused by these spurious ORFs (Das et al., 1997).
Structures were taken from the PDB via the PDB browser [67, 90]. Domain fold and class definitions were taken from scop (version 1.35, May 1996) [9, 58, 91]. Specific values quoted about the composition of the PDB, e.g. that it has 5493 total structures and 222 T4 lysozyme structures, refer to the state of the databank when scop 1.35 was built. Core structures for each domain were based on refinement of structural alignments [12, 30, 92, 93]. only in a} The biophysical protein list was constructed in a subjective fashion, based on conversations with colleagues and reading the literature.
Analysis and processing of the data was greatly expedited by the use of a simple relational database, implemented in DBM, Perl5 [94] and mini-SQL (http://Hughes.com.au). This was described previously [12]. It has tables cross-referencing sequence identifiers, structure matches, TM-helix positions, and so forth and cross-tabulation reports giving the occurrence of various patterns. Most of these tables and reports will be made available over the Internet (as text tables and via a simple query interface) from the following URL: http://bioinfo.mbb.yale.edu/genome. The tables are structured in such a way that all the genome features (e.g. location of a TM-helix or PDB match) are annotated in a consistent fashion, with thresholds and scoring schemes applied consistently over multiple tools. This attempt at consistency is similar to what has been achieved in other genome annotation systems that aim to integrate multiple tools [29, 95].
Matching to Known Structures
All sequence comparison was done with the FASTA program (version 2.0) with k-tup 1 and an "e-value" threshold of 0.01 [96, 97]. The e-value describes the number of false positives expected in a single database scan, so a value of 0.01 means that about one out of a hundred cluster linkages will be in error [9, 98-102]. This error rate has been verified by empirical tests on a database of known protein relationships and is similar to the thresholds used in other multi-genome comparisons [9, 16, 98]. Probabilistic scores, such as the e-value, should give similar results to more conventional scores, such as percent identity, but they have been shown to be better calibrated and more sensitive for marginal similarities, taking into account compositional biases of the databank and the query sequence [99, 102-104].
There are other, potentially more sensitive, methods of comparing sequences to structures than FASTA, e.g. profiles, Hidden-Markov models, motif analysis, secondary structure matching, and threading [105-110]. A number of these were tested, and as expected, they find more homologues for certain folds. However, the sensitivity improvement is not uniform over all folds. This is not advantageous for a large-scale census where uniform sampling and treatment of the data is more important than sensitivity. In this instance one is more concerned with accurate relative numbers than with absolute values. Cobbling together a census through the use of a disparate collection of tools and patterns creates the problem of devising consistent scores and thresholds. This is particularly acute in the case of manually derived sequence patterns and motifs, since an expert on a particular fold or motif would expect his pattern to find relatively more homologues than a pattern not constructed by an expert. The approach here, applying the same objective procedure to each fold, circumvents these problems to some degree. Furthermore, it has the added advantage that it can be performed automatically without manual intervention and, consequently, can easily be scaled up to deal with much larger data sets.
Another issue to consider with regard to matching sequences to structures has to do with the fact that protein structure is fundamentally arranged around the level of folding domains whereas statistics for genomes are often calculated and best understood in terms of the number of genes. For instance, when one talks about how prevalent the kinase and Rossmann folds are in the yeast and E. coli genomes, one is implicitly comparing the number of matches that known kinase and Rossmann fold structures have in the ~6200 yeast ORFs relative to the ~4300 E. coli ORFs. However, it is possible for a single gene to contain a number of kinase fold domains or to simultaneously contain both a kinase and Rossmann fold. Thus, the total number of domains in a genome is probably a better standard for these comparisons. Unfortunately, this number is not known. But it is known that the number of domains is not related simply to the number of genes. For instance, on average a protein is about 50% larger in yeast than in E. coli (317 vs. 466), meaning that there are probably twice as many possible domains in yeast as in E. coli. Here an intermediate approach is taken. The statistics are reported in terms of the number of domains matched but reference is always made to the number of ORFs in the genome.
Clustering and Trees
The structures in the PDB were clustered into 990 representative domains. The few membrane protein structures in the PDB were excluded from this clustering so that all the membrane proteins would be identified, in a uniform fashion, by prediction. (This is not expected to be a major factor as, for instance, the yeast genome contains only a single homologue to a known membrane protein structure). The clustering was similar in spirit to the many previous divisions of the PDB into representative chains (e.g. [68, 98, 111-113]). However, a slightly different multiple-linkage algorithm was used [114]. It was designed to be internally consistent with the search method used to identify homologues in the genomes, using the same similarity criteria (a FASTA e-value threshold). The clustering algorithm takes the results of an all-vs-all comparison of the PDB and creates a graph that has one vertex for each sequence and one edge for each similarity score. Each vertex starts out as a cluster of size one. Since sequence similarity scores (i.e. e-values) are not commutative, this directed graph is converted to an undirected graph by removing the better scoring edges between pairs. Then, each edge is considered in turn, and the two clusters associated by this edge are merged into a single cluster if every member of the first cluster has a good scoring edge between it and every member of the second cluster, and vice versa. The edges are considered in order of decreasing similarity. This has the advantage that close relationships are considered before more distant ones, ensuring that distant relationships are not erroneously used to add a member to a cluster when there exists (for that member) a much closer relationship that would lead to an alternate clustering. Furthermore, this algorithm will produce the same result on the same data set every time; i.e. it is not affected by the order in which the data is traversed.
Trees based on distance matrices were built with simple UPGMA clustering using the Kitsch program, which is part of the Phylip package [115, 116]. Trees were built on the basis of the difference in amino acid composition vectors, as described in the caption to Figure 3. Di-amino acid composition was also used and gave a similar tree.
After the clustering was completed, it was found that the PDB consisted of 990 non-homologous domains, each of which represents a single sequence family. These 990 domains were grouped into 338 fold families by the structural relationships in scop [58]. Each of the 338 folds can be ranked in terms of how many of the 990 sequence families it contains. It was decided to define a superfold as one of the top 25 folds in terms of the number of associated sequence families. Each of these contains at least ten sequence families. This threshold is arbitrary and is similar but not identical to past usage [59].
Transmembrane Helix and Low-Complexity Region Identification
Transmembrane segments were identified using the GES hydrophobicity scale [117], [Engelman, 1986 #1224]. The values from the scale for amino acids in a window of size 20 (the typical size of a transmembrane helix) were averaged and then compared against a cutoff of -1 kcal/mole. A value under this cutoff was taken to indicate the existence of a transmembrane helix. Initial hydrophobic stretches corresponding to signal sequences for membrane insertion were excluded. (These have the pattern of a charged residue within the first seven, followed by a stretch of 14 with an average hydrophobicity under the cutoff.) These parameters have been used, tested, and refined on surveys of membrane protein in genomes [50, 53, 55].
Low-complexity sequences were identified with the SEG program [65, 118, 119] using the standard parameters K(1)= 3.4 and K(2)=3.75, and a window of length 45. These parameters are the ones used to find "long" domain-size low-complexity regions. The average size of a low-complexity region found here is ~110 residues. Many of these transmembrane regions are also low complexity regions (almost half). Taking a conservative approach, it was decided to annotate these doubly identified regions as low complexity, not as transmembrane. This will tend to reduce the total amount of identified TM-helices. This is especially true for MJ, which has the largest amount of low-complexity regions.
Acknowledgements
Thanks to Guy Plunket and Mike Cherry, for providing information about the genome data; Steve Chervitz, for information about Sacch3D; George Weinstock and Steve Norris, for providing information on transmembrane folds; Fred Richards, Lynne Regan, and Julie Forman-Kaye for helping with the biophysical protein lists; Hedi Hegyi, for carefully reading the manuscript; Ted Johnson, for help in structure clustering; and Janice Murphy, for manuscript preparation.
References
1. Nowak, R. Bacterial Genome Sequence Bagged. Science 269: 468-470, 1995.
2. Langreth, R. Scientists Unlock Sequence Of Ulcer Bacterium's Genes. Wall Street Journal B1, 1997.
3. Wade, N (1997). Thinking Small Paying Off Big In Gene Quest. New York Times, 3 February 1997, A1 (front page).
4. Blaisdell, B E, Campbell, A M & Karlin, S. Similarities and dissimilarities of phage genomes. Proceedings of the National Academy of Sciences of the United States of America 93: 5854-5859, 1996.
5. Karlin, S & Burge, C. Dinucleotide relative abundance extremes: a genomic signature. [Review]. Trends in Genetics 11: 283-290, 1995.
6. Karlin, S, Burge, C & Campbell, A M. Statistical analyses of counts and distributions of restriction sites in DNA sequences. Nucleic Acids Research 20: 1363-1370, 1992.
7. Karlin, S, Mrazek, J & Campbell, A M. Frequent oligonucleotides and peptides of the haemophilus influenzae genome. Nucleic Acids Research 24: 4263-4272, 1996.
8. Koonin, E V, Mushegian, A R & Rudd, K E. Sequencing and analysis of bacterial genomes. Curr Biol 6: 404-416, 1996.
9. Brenner, S, Hubbard, T, Murzin, A & Chothia, C. Gene Duplication in H. Influenzae. Nature 378: 140, 1995.
10. Riley, M. Genes and proteins of Escherichia coli K-12 (GenProtEC). Nucleic Acids Res 25: 51-52, 1997.
11. Wolfe, K H & Shields, D C. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387: 708-713, 1997.
12. Gerstein. A Structural Census of Genomes: Comparing Eukaryotic, Bacterial and Archaeal Genomes in terms of Protein Structure. J. Mol. Biol. 274: 562-576, 1997.
13. Tamames, J, Casari, G, Ouzounis, C & Valencia, A. Conserved clusters of functionally related genes in two bacterial genomes. J Mol Evol 44: 66-73, 1997.
14. Green, P, Lipman, D, Hillier, L, Waterston, R, States, D & Claverie, J M. Ancient conserved regions in new gene sequences and the protein databases. Science 259: 1711-1716, 1993.
15. Koonin, E V, Tatusov, R L & Rudd, K E. Sequence similarity analysis of Escherichia coli proteins: Functional and Evolutionary Implications. Proc. Natl. Acad. Sci. USA 92: 11921-11925, 1995.
16. Tatusov, R L, Koonin, E V & Lipman, D J. A genomic perspective on protein families. Science 278: 631-637, 1997.
17. Ouzounis, C, Kyrpides, N & Sander, C. Novel protein families in Archaean genomes. Nucl. Acids Res. 23: 565-570, 1995.
18. Ouzounis, C, Bork, P, Casari, G & Sander, C. New protein functions in yeast chromosome VIII. Protein Sci. 4: 2424-2428, 1995.
19. Clayton, R A, White, O, Ketchum, K A & Venter, J C. The first genome from the third domain of life [news]. Nature 387: 459-462, 1997.
20. Karp, P, Riley, M, Paley, S & Pellegrini-Toole, A. EcoCyc: Electronic Encyclopedia of E. coli Genes and Metabolism. Nucleic Acids Research 24: 32-40, 1996.
21. Karp, P D, Ouzounis, C & Paley, S M (1996). HinCyc: A knowledge base of the complete genome and metabolic pathways of H. influenze, in Proc. Fourth Intl. Conf. Intell. Sys. Mol. Biol. 116-124 (AAAI Press, Menlo Park,
22. Mushegian, A R & Koonin, E V. A minimal gene set for cellular life derived by comparison of complete bacterial genomes [see comments]. Proc Natl Acad Sci U S A 93: 10268-10273, 1996.
23. Tatusov, R L, et al. Metabolism and evolution of Haemophilus influenzae deduced from a whole- genome comparison with Escherichia coli. Curr Biol 6: 279-291, 1996.
24. Fleischmann, R D, et al. Whole-genome random sequencing and assembly of haemophilus influenzae rd. Science (Washington D C) 269: 496-512, 1995.
25. Bork, P, Ouzounis, C, Sander, C, Scharf, M, Schneider, R & Sonnhammer, E. What's in a genome? Nature 358: 287, 1992.
26. Bork, P, Ouzounis, C, Sander, C, Scharf, M, Schneider, R & Sonnhammer, E. Comprehensive sequence analysis of the 182 predicted open reading frames of yeast chromosome iii. Protein Science 1: 1677-1690, 1992.
27. Scharf, M, Schneider, R, Casari, G, Bork, P, Valencia, A, Ouzounis, C & Sander, C (1994). GeneQuiz: a workbench for sequence analysis, in Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology. 348-353 (AAAI Press, Menlo Park, California).
28. Casari, G, et al. Challenging times for bioinformatics. Nature 376: 647-648, 1995.
29. Gaasterland, T & Sensen, C W. Fully automated genome analysis that reflects user needs and preferences. A detailed introduction to the MAGPIE system architecture. Biochimie 78: 302-310, 1996.
30. Gerstein, M & Altman, R. Average core structures and variability measures for protein families: Application to the immunoglobulins. J. Mol. Biol. 251: 161-175, 1995.
31. Gerstein, M & Altman, R. A Structurally Invariant Core for the Globins. CABIOS 11: 633-644, 1995.
32. Henikoff, S & Henikoff, J G. Automated assembly of protein blocks for database searching. Proc. Natl. Acad. Sci. 19: 6565-6572, 1993.
33. Henikoff, S & Henikoff, J G. Protein family classification based on searching a database of blocks. Genomics 19: 97-107, 1994.
34. Henikoff, S, Greene, E A, Pietrokovski, S, Bork, P, Attwood, T K & Hood, L. Gene families: the taxonomy of protein paralogs and chimeras. Science 278: 609-614, 1997.
35. Sonnhammer, E L L & Kahn, D. Modular arrangement of proteins as inferred from analysis of homology. Protein Science 3: 482-492, 1994.
36. Sonnhammer, E, Eddy, S & Durbin, R. Pfam: a Comprehensive Database of Protein Domain Families Based on Seed Alignments. Proteins 28: 405-420, 1997.
37. Riley, M & Labedan, B. Protein evolution viewed through Escherichia coli protein sequences: introducing the notion of a structural segment of homology, the module. J Mol Biol 268: 857-868, 1997.
38. Fabian, P, Murvai, J, Hatsagi, Z, Vlahovicek, K, Hegyi, H & Pongor, S. The SBASE protein domain library, release 5.0: a collection of annotated protein sequence segments. Nucleic Acids Res 25: 240-243, 1997.
39. Chothia, C & Lesk, A M. The relation between the divergence of sequence and structure in proteins. EMBO J. 5: 823-826, 1986.
40. Chothia, C & Gerstein, M. Protein evolution. How far can sequences diverge? Nature 385: 579-581, 1997.
41. Chothia, C. Proteins — 1000 families for the molecular biologist. Nature 357: 543-544, 1992.
42. Lander, E S. The Genomics: Global Views of Biology. Science 274: 536-539, 1996.
43. Lesk, A M, Levitt, M & Chothia, C. Alignment of amino acid sequences of distantly related proteins using variable gap penalties. Prot. Eng. 1: 77-78, 1986.
44. Bork, P, Ouzounis, C & Sander, C. From Genome Sequences to Protein Function. Curr. Opin. Struct. Biol. 4: 393-403, 1994.
45. Gerstein, M & Levitt, M. A Structural Census of the Current Population of Protein Sequences. Proc. Natl. Acad. Sci. USA 94: 11911-11916, 1997.
46. Fischer, D & Eisenberg, D. Assigning folds to the proteins encoded by the genome of mycoplasma genitalium [In Process Citation]. Proc Natl Acad Sci U S A 94: 11929-11934, 1997.
47. Goffeau, A, Slonimski, P, Nakai, K & Risler, J L. How Many Yeast Genes Code for Membrane-Spanning Proteins? Yeast 9: 691-702, 1993.
48. Rost, B, Fariselli, P, Casadio, R & Sander, C. Prediction of helical transmembrane segments at 95% accuracy. Prot. Sci. 4: 521-533, 1995.
49. Rost, B. PHD: Predicting One-dimensional Protein Secondary Structure by Profile-Based Neural Networks. Meth. Enz. 266: 525-539, 1996.
50. Arkin, I, Brunger, A & Engelman, D. Are there dominant membrane protein families with a given number of helices? Proteins 28: 465-466, 1997.
51. Boyd, D, Schierle, C & Beckwith, J. How many membrane proteins are there? Prot. Sci. 7: 201-205, 1998.
52. Jones, D T. Do transmembrane protein superfolds exist? FEBS Lett 423: 281-285, 1998.
53. Tomb, J-F, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388: 539-547, 1997.
54. Fraser, C M, et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi [see comments]. Nature 390: 580-586, 1997.
55. Wallin, E & von Heijne, G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms [In Process Citation]. Protein Sci 7: 1029-1038, 1998.
56. Gibrat, J F, Madej, T & Bryant, S H. Surprising similarities in structure comparison. Curr. Opin. Str. Biol. 6: 377-385, 1996.
57. Holm, L & Sander, C. Mapping the Protein Universe. Science 273: 595-602, 1996.
58. Murzin, A, Brenner, S E, Hubbard, T & Chothia, C. SCOP: A Structural Classification of Proteins for the Investigation of Sequences and Structures. J. Mol. Biol. 247: 536-540, 1995.
59. Orengo, C A, Jones, D T & Thornton, J M. Protein superfamilies and domain superfolds. Nature 372: 631-634, 1994.
60. Schmidt, R, Gerstein, M & Altman, R. LPFC: An Internet Library of Protein Family Core Structures. Prot. Sci. 6: 246-248, 1997.
61. Pascarella, S & Argos, P. A Databank Merging Related Protein Structures and Sequences. Prot. Eng. 5: 121-137, 1992.
62. Sander, C & Schneider, R. Database of Homology-Derived Protein Structures and the Structural Meaning of Sequence Alignment. Proteins: Struc. Func. Genet. 9: 56-68, 1991.
63. Orengo, C A, Michie, A D, Jones, S, Jones, D T, Swindells, M B & Thornton, J M. CATH--a hierarchic classification of protein domain structures. Structure 5: 1093-1108, 1997.
64. Weiss, M S, Abele, U, Weckesser, J, Welte, W, Schiltz, E & Schulz, G E. Molecular architecture and electrostatic properties of a bacterial porin. Science 254: 1627-1630, 1991.
65. Wootton, J C & Federhen, S. Analysis of compositionally biased regions in sequence databases. Methods Enzymol 266: 554-571, 1996.
66. Champion, C I, et al. Sequence analysis and recombinant expression of a 28-kilodalton Treponema pallidum subsp. pallidum rare outer membrane protein (Tromp2). J Bacteriol 179: 1230-1238, 1997.
67. Abola, E, Sussman, J, Prilusky, J & Manning, N. Protein Data Bank archives of three-dimensional macromolecular structures. Meth. Enz. 277: 556-571, 1997.
68. Brenner, S E, Chothia, C & Hubbard, T J. Population statistics of protein structures: lessons from structural classifications [In Process Citation]. Curr Opin Struct Biol 7: 369-376, 1997.
69. Doolittle, R F. The multiplicity of domains in proteins. [Review]. Annual Review of Biochemistry 64: 287-314, 1995.
70. Govindarajan, S & Goldstein, R A. Why are some proteins structures so common? Proc Natl Acad Sci U S A 93: 3341-3345, 1996.
71. Li, H, Helling, R, Tang, C & Wingreen, N. Emergence of preferred structures in a simple model of protein folding [see comments]. Science 273: 666-669, 1996.
72. Sonnhammer, E L & Durbin, R. Analysis of protein domain families in Caenorhabditis elegans. Genomics 46: 200-216, 1997.
73. Ouzounis, C & Kyrpides, N. The emergence of major cellular processes in evolution. FEBS Lett 390: 119-123, 1996.
74. Sneath, P H A & Sokal, R R (1973). Numerical Taxonomy (W H Freeman, San Francisco).
75. Olsen, G J, Woese, C R & (1994)., R O. J. Bacteriol. 176: 1-6, 1994.
76. Doolittle, R F. A bug with excess gastric avidity [news; comment]. Nature 388: 515-516, 1997.
77. Levitt, M & Chothia, C. Structural patterns in globular proteins. Nature 261: 552-558, 1976.
78. Velculescu, V E, et al. Characterization of the yeast transcriptome. Cell 88: 243-251, 1997.
79. Lashkari, D A, et al. Yeast microarrays for genome wide parallel genetic and gene expression analysis. Proc Natl Acad Sci U S A 94: 13057-13062, 1997.
80. DeRisi, J L, Iyer, V R & Brown, P O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278: 680-686, 1997.
81. Knuth, D (1973). The Art of Computer Programming: vol 3, Sorting and Searching (Addison-Wesley, Reading, MA).
82. Konopka, A K & Martindale, C. Noncoding DNA, Zipf's law, and language [letter]. Science 268: 789, 1995.
83. Flam, F. Hints of a language in junk DNA [news] [see comments]. Science 266: 1320, 1994.
84. Bornberg-Bauer, E. How are model protein structures distributed in sequence space? [In Process Citation]. Biophys J 73: 2393-2403, 1997.
85. Cherry, J M, et al. SGD: Saccharomyces Genome Database. Nucleic Acids Res 26: 73-79, 1998.
86. Kerlavage, A R. TIGR Microbial Genome Database. http://www.tigr.org/mdb (as of 2/97), 1997.
87. Blattner, F R, et al. The Complete Genome Sequence of Escherichia coli K-12. Science 277: 1453-1462, 1997.
88. Goffeau, A, et al. Life with 6000 Genes. Science 274: 546-567, 1996.
89. Kim, J M, Vanguri, S, Boeke, J D, Gabriel, A & Voytas., D F. Transposable elements and genome organization: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Research In press, 1998.
90. Stampf, D R, Felder, C E & Sussman, J L. PDBbrowse--a graphics interface to the Brookhaven Protein Data Bank. Nature 374: 572-574, 1995.
91. Hubbard, T J P, Murzin, A G, Brenner, S E & Chothia, C. SCOP: a structural classification of proteins database. Nucleic Acids Res 25: 236-239, 1997.
92. Altman, R & Gerstein, M (1994). Finding an Average Core Structure: Application to the Globins, in Proceedings of the Second International Conferene on Intelligent Systems in Molecular Biology. 19-27 (AAAI Press, Menlo Park, CA).
93. Gerstein, M & Levitt, M (1996). Using Iterative Dynamic Programming to Obtain Accurate Pairwise and Multiple Alignments of Protein Structures, in Proc. Fourth Int. Conf. on Intell. Sys. Mol. Biol. 59-67 (AAAI Press, Menlo Park, CA).
94. Wall, L, Christiansen, D & Schwartz, R (1996). Programming Perl (O'Reilly and Associates, Sebastapol, CA).
95. Medigue, C, Moszer, I, Viari, A & Danchin, A. Analysis of a Bacillus subtilis genome fragment using a co-operative computer system prototype. Gene 165: GC37-51, 1995.
96. Lipman, D J & Pearson, W R. Rapid and sensitive protein similarity searches. Science 227: 1435-1441, 1985.
97. Pearson, W R & Lipman, D J. Improved Tools for Biological Sequence Analysis. Proc. Natl. Acad. Sci. USA 85: 2444-2448, 1988.
98. Brenner, S, Chothia, C & Hubbard, T. Assessing sequence comparison methods with reliable structurally identified distant evolutionary relationships. Proc. Natl. Acad. Sci. USA 95: 6073-6078, 1998.
99. Pearson, W R. Empirical statistical estimates for sequence similarity searches. J Mol Biol 276: 71-84, 1998.
100. Pearson, W R. Effective Protein Sequence Comparison. Meth. Enz. 266: 227-259, 1996.
101. Pearson, W R. Identifying distantly related protein sequences. Comput Appl Biosci 13: 325-332, 1997.
102. Levitt, M & Gerstein, M. A Unified Statistical Framework for Sequence Comparison and Structure Comparison. Proceedings of the National Academy of Sciences USA 95: 5913-5920, 1998.
103. Altschul, S F, Boguski, M S, Gish, W & Wootton, J C. Issues in searching molecular sequence databases. [Review]. Nature Genetics 6: 119-129, 1994.
104. Karlin, S & Altschul, S F. Applications and statistics for multiple high-scoring segments in molecular sequences. Proceedings of the National Academy of Sciences of the United States of America 90: 5873-5877, 1993.
105. Bowie, J U & Eisenberg, D. Inverted protein structure prediction. Curr Opin Struct Biol 3: 437-444, 1993.
106. Jones, S & Thornton, J. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 93: 13-20, 1996.
107. Eddy, S R. Hidden Markov models. Curr. Opin. Struc. Biol. 6: 361-365, 1996.
108. Tatusov, R L, Altschul, S F & Koonin, E V. Detection of conserved segments in proteins: iterative scanning of sequence databases with alignment blocks. Proc Natl Acad Sci U S A 91: 12091-12095, 1994.
109. Dubchak, I, Muchnik, I, Holbrook, S R & Kim, S H. Prediction of protein folding class using global description of amino acid sequence. Proc Natl Acad Sci U S A 92: 8700-8704, 1995.
110. Aurora, R & Rose, G D. Seeking an ancient enzyme in Methanococcus jannaschii using ORF, a program based on predicted secondary structure comparisons. Proc Natl Acad Sci U S A 95: 2818-2823, 1998.
111. Hobohm, U, Scharf, M, Schneider, R & Sander, C. Selection of a representative set of structures from the Brookhaven Protein Data Bank. Protein Science 1: 409-417, 1992.
112. Hobohm, U & Sander, C. Enlarged representative set of protein structures. Protein Science 3: 522, 1994.
113. Boberg, J, Salakoski, T & Vihinen, M. Selection of a representative set of structures from Brookhaven Protein Data Bank. Proteins 14: 265-276, 1992.
114. Kaufman, L & Rousseeuw, P J (1990). Finding Groups in Data: An Introduction to Cluster Analysis (John Wiley & Sons, New York).
115. Felsenstein, J. PHYLIP — Phylogeny Inference Package (Verstion 3.2). Cladistics 5: 164-166, 1989.
116. Felsenstein, J (1993). PHYLIP (Phylogeny Inference Package) version 3.5c., (Department of Genetics, University of Washington, Seattle).
117. Engelman, D M, Steitz, T A & Goldman, A. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. [Review]. Annual Review of Biophysics & Biophysical Chemistry 15: 321-353, 1986.
118. Wootton, J C & Federhen, S. Statistics of local complexity in amino acid sequences and sequence databases. Computers and Chemistry 17: 149-163, 1993.
119. Wootton, J C. Non-globular domains in protein sequences: automated segmentation using complexity measures. Comput Chem 18: 269-285, 1994.
120. Fraser, C M, et al. The minimal gene complement of Mycoplasma genitalium [see comments]. Science 270: 397-403, 1995.
121. Bult, C J, et al. Complete Genome Sequence of the Methanogenic Archaeon, Methanococcus jannaschii. Science 273: 1058-1073, 1996.
122. Himmelreich, R, Hilbert, H, Plagens, H, Pirkl, E, Li, B C & Herrmann, R. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res 24: 4420-4449, 1996.
123. Goffeau, A & names], e a. The Yeast Genome Directory. Nature 387(Supp): 5-105, 1997.
124. Kaneko, T, et al. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res 3: 109-136, 1996.
125. Amari, S. Annals of Statistics 10: 357-387, 1982.
126. Kraulis, P J. MOLSCRIPT - A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24: 946-950, 1991.
Table 1, Genomes and Abbreviations Used
|
Abbrev. EC |
Bacteria (gram negative) |
Escherichia coli |
4.60 |
4290 |
[87] |
www.genetics.wisc.edu |
||||||
|
HI |
Bacteria (gram negative) |
Haemophilus influenzae |
1.83 |
1680 |
[24] |
www.tigr.org/tdb/mdb/hidb/hidb.html |
||||||
|
HP |
Bacteria (gram negative) |
Helicobacter pylori |
1.66 |
1577 |
[53] |
www.tigr.org/tdb/mdb/hpdb/hpdb.html |
||||||
|
MG |
Bacteria (gram positive) |
Mycoplasma genitalium |
0.58 |
468 |
[120] |
www.tigr.org/tdb/mdb/mgdb/mgdb.html |
||||||
|
MJ |
Archaea (Euryarchaeota) |
Methanococcus jannaschii |
1.66 |
1735 |
[121] |
www.tigr.org/tdb/mdb/mjdb/mjdb.html |
||||||
|
MP |
Bacteria (gram positive) |
Mycoplasma pneumoniae |
0.81 |
677 |
[122] |
www.zmbh.uni-heidelberg.de/ |
||||||
|
SC |
Eukarya (fungi) |
Saccharomyces cerevisiae |
13 |
6218 |
[123] |
Genome-www.stanford.edu/ |
||||||
|
SS |
Bacteria (Cyanobacteria) |
Synechocystis sp. |
3.57 |
3168 |
[124] |
www.kazusa.or.jp/cyano/cyano.html |
||||||
Table 2, The Observed Patterns of Fold Usage
E
SHSHMMM (##) ESHSHMMM (##) ESHSHMMM (##) ESHSHMMM (##) ESHSHMMM (##)CCISPJPG CCISPJPG CCISPJPG CCISPJPG CCISPJPG
11111111 (30) .1...... (23) 1....... (19) 11111.11 (16) 111111.. (16)
1111.... (09) 11111... (08) 1.1..... (08) 1.111.11 (06) 11...... (06)
...1.... (06) 1.11.... (05) .1.1.... (05) 1.111... (04) 11.1.... (04)
.1...1.. (04) ..1..... (04) 111111.1 (03) 1111111. (03) 1111..11 (03)
1111.1.. (03) .....1.. (03) 1111.111 (02) 111...11 (02) 111.11.. (02)
1.11.1.. (02) ..111... (02) .1.11... (02) 1..1.1.. (02) 1.1..1.. (02)
111..... (02) .11..... (02) ......1. (02) ....1... (02) 111..111 (01)
111.1.11 (01) 1.111..1 (01) 1.1111.. (01) .1.1..11 (01) .1.11.1. (01)
.11.1..1 (01) 1....111 (01) 1..111.. (01) 1.1...11 (01) 1.1..11. (01)
11....11 (01) 11.1.1.. (01) 11.11... (01) 111..1.. (01) 111.1... (01)
.11...1. (01) 1.....11 (01) 1...11.. (01) 1.1.1... (01) ......11 (01)
....1..1 (01) ...1.1.. (01) ...11... (01) ..1.1... (01) .1....1. (01)
1....1.. (01) .......1 (01)
Each of the 8-bit binary numbers represents a particular pattern of fold usage: "1" if a fold is present and "." if it is absent. From left to right, the bits correspond to EC, SC, HI, SS, HP, MJ, MP, and MG. The number in parentheses after the binary number is the number of folds that have this pattern of usage. For instance, "..1..... (04)" means that there are four folds present in HI and in no other of the genomes. There are 255 possible patterns of fold usage, but only the 62 that are observed are shown here.
Table 3, Folds Present in or Absent from Only a Single Genome
|
Overall Totals |
Number of Times Only from This Genome |
Number of Times Only in This Genome |
|||||||||||
|
matches to the PDB |
distinct folds |
fold |
fam. |
all- a |
all- b |
fold |
fam. |
all- a |
all- b |
||||
|
EC |
848 |
174 |
. |
. |
. |
. |
19 |
51 |
1 |
6 |
|||
|
SC |
1073 |
157 |
. |
3 |
. |
. |
23 |
84 |
5 |
3 |
|||
|
HI |
394 |
146 |
. |
. |
. |
. |
4 |
11 |
. |
1 |
|||
|
SS |
534 |
140 |
. |
1 |
. |
. |
6 |
24 |
. |
3 |
|||
|
HP |
254 |
107 |
2 |
4 |
. |
1 |
2 |
11 |
. |
1 |
|||
|
MJ |
233 |
82 |
16 |
29 |
4 |
3 |
3 |
6 |
1 |
1 |
|||
|
MP |
140 |
76 |
3 |
3 |
. |
1 |
2 |
3 |
1 |
. |
|||
|
MG |
134 |
74 |
3 |
1 |
. |
. |
1 |
2 |
1 |
. |
|||
|
total |
3610 |
338 |
24 |
41 |
4 |
5 |
60 |
192 |
9 |
15 |
|||
Column 2 ("matches") shows how many total homologues there are for a particular genome to structures in the PDB (i.e. total PDB hits in the genome). Column 3 ("distinct folds") shows how many different folds are contained in this genome, regardless of whether these folds occur in other genomes. Columns 4 to 7 (under "ABSENT") show how many times one of the 338 folds, 990 sequence families, 48 all-alpha folds, or 39 all-beta folds are absent only from this genome (and not from the other seven). Columns 8 to 11 ("PRESENT"), conversely, show how many times a fold or family is ONLY present in this genome, i.e. how many unique folds the genome has.
Table 4, Yeast Folds Ranked by Duplication and Expression
|
Common Yeast Folds (scop) |
Rep. Structure |
Genome Duplication |
Expression (aerobic) |
Expression (anaerobic) |
|
Protein kinases (cat. core) |
1hcl |
1 |
3 |
4 |
|
NTP Hydrolases with P-loop |
1gky |
2 |
1 |
2 |
|
Classic Zn finger |
1ard |
3 |
9 |
5 |
|
Ribonuclease H-like motif |
2rn2 |
4 |
2 |
1 |
|
Rossmann Fold |
1xel |
5 |
4 |
3 |
|
Zn2/Cys6 DNA-binding dom. |
125d |
6 |
6 |
7 |
|
7-bladed beta-propeller |
2bbk-H |
7 |
8 |
16 |
|
TIM-barrel |
1byb |
8 |
5 |
6 |
|
like Ferrodoxin |
1fxd |
9 |
7 |
10 |
|
DNA-binding 3-helix bundle |
1enh |
10 |
30 |
36 |
|
… |
… |
|||
|
GroES-like |
1lep-A |
17 |
10 |
9 |
|
… |
… |
|||
|
like HSP70, Ct-dom. |
1dkz-A |
22 |
11 |
8 |
This table shows the most common folds in the yeast genome ranked according to duplication and expression. Column 1 and 2 ("name" and "Rep. Struc.") gives the name for the fold, as determined by scop [58], and a representative structure with this fold. (In the table "dom" is used as an abbreviation for domain, "Nt-dom," for N-terminal domain, and "Ct-dom," for C-terminal domain.) Column 3 ("Duplication") gives an ordering of folds in terms of the number of times they are found in the yeast genome. For instance, the top fold (kinase) is found 110 times, while the second fold (NTP hydrolase) is found 69 times (from data in the figure 1 fold table). Columns 4 and 5 ("expression") give an ordering of folds in terms of their degree of expression. Using the data from DeRisi et al. [80], the total expression E of a fold F is calculated as a sum of the expression levels of all the ORFs that contain this fold. The expression level of a given ORF (i.e. ORF i) is the degree of its "Red" color on a cDNA microarray R(i), less the background Rback(i), viz: ![]() Column 4 gives the expression in aerobic conditions (high sugar, second time-series data point in DeRisi et al.), and Column 5, in anaerobic conditions (low sugar, high ethanol, last time-series data point in DeRisi et al.). Note how some folds that are in the top-10 in terms of duplication are not in this select list in terms of expression (e.g. "DNA-binding 3-helical bundle").
Column 4 gives the expression in aerobic conditions (high sugar, second time-series data point in DeRisi et al.), and Column 5, in anaerobic conditions (low sugar, high ethanol, last time-series data point in DeRisi et al.). Note how some folds that are in the top-10 in terms of duplication are not in this select list in terms of expression (e.g. "DNA-binding 3-helical bundle").
Table 5, The Yeast Top-10 Determined Two Ways
|
Common Yeast Folds |
Rep. Struct. |
GeneCensus |
Sacch3D, SGD |
diff. |
||||
|
count |
rank |
count |
rank |
|||||
|
Protein kinases (cat. core) |
1hcl |
110 |
1 |
109 |
1 |
|||
|
NTP Hydrolases with P-loop |
1gky |
69 |
2 |
52 |
2 |
|||
|
Classic Zn finger |
1ard |
55 |
3 |
34 |
7 |
. |
||
|
Ribonuclease H-like motif |
2rn2 |
54 |
4 |
30 |
8 |
. |
||
|
Rossmann Fold |
1xel |
46 |
5 |
41 |
5 |
|||
|
Zn2/Cys6 DNA-binding dom. |
125d |
46 |
6 |
30 |
9 |
. |
||
|
7-bladed beta-propeller |
2bbk-H |
46 |
7 |
0 |
- |
< |
||
|
TIM-barrel |
1byb |
36 |
8 |
39 |
6 |
. |
||
|
Ferrodoxin-like |
1fxd |
28 |
9 |
43 |
4 |
. |
||
|
DNA-binding 3-helix bundle |
1enh |
22 |
10 |
22 |
10 |
|||
|
Long Helix Oligomers (coils) |
1zta |
1 |
- |
47 |
3 |
< |
||
This table shows the top-10 folds in yeast calculated two ways, as done here (GeneCensus) versus as done for the Sacch3D database, which is part of SGD [85]. Columns 1 and 2 ("name" and "Rep. Struc.") give the name for the fold, as determined by scop [58], and a representative structure with this fold. (In the table "dom" is used as an abbreviation for domain, "Nt-dom," for N-terminal domain, and "Ct-dom," for C-terminal domain.) The columns labeled "GeneCensus" show the top-10 folds determined with the methods used here: FASTA with e-value cutoff of .01 and strict overlap criteria, using a clustered version of the scop 1.35 database. The columns labeled by "Sacch3D" show the top-10 folds calculated by different methods: WU-BLAST with a p-value cutoff of 10e-4, using a differently clustered version of an earlier scop database, 1.32. For both GeneCensus and Sacch3D both the number of folds found ("count") and the rank in the top-10 list are shown. There is broad agreement between the two top-10 lists. However, there are some differences. These are flagged in the final column. The minor differences (indicated by ".") are the five folds that have slightly changed rank in within the top-10. In comparison to BLAST, FASTA appears to be a bit better in finding homologues for the Classic Zn finger, Ribonuclease H-like motif, and the Rossmann Fold and a bit worse for the TIM-barrel and Ferrodoxin-like folds. The two major differences (indicated by "<") are more substantial and warrant explanation:
(1) Propeller Fold. The Sacch3D list does not have any "7-bladed beta-propeller" folds. This is because of new structures that were added to scop between release 1.32 and 1.35, in particular the structure of transducin. This difference, thus, illustrates how the top-10 lists evolve with the growth of the PDB.
(2) Helix Oligomers. The Sacch3D list has many more long helix oligomer folds. These are small "folds" such as coiled-coils and leucine zippers. These undoubtedly occur in yeast in great frequency. However, they are very small in size -- e.g. the representative coiled-coil d2ztaa_ is only 30 residues. This makes them particularly problematical to define as a fold and to find with sequence searching programs. The difference between the BLAST and FASTA results illustrates dramatically how certain programs may differ in finding these marginal folds.
Figure 1 (caption), Fold Usage in Different Genomes
This table shows the usage of each of the known folds in eight different genomes. The entire table is available over the web at http://bioinfo.mbb.yale.edu/genome/browser/fold-report. Here only the top part of the table is shown, corresponding to the folds that occur in at least seven genomes. In all columns inverted (white-on-black) squares are for numbers >10, gray squares are for 2 to 9, and white squares are for 1. Column 1 ("class") is the structural class that the fold belongs to, as determined by scop [58]. Column 2 ("Fold#") is the fold number in scop 1.35. Columns 3 to 10 ("EC" to "MG") give the total number of matches in one genome for a particular fold. This is on a domain level so there can potentially be more than one match per ORF (see text). For instance, the first row shows that there are 19 Rossmann fold domains in the HP genome. The columns are sorted in terms of the total number of matches in the genome with EC having the most and MG, the least. Column 11 ("Total") is the row total of columns 3 to 10, the total number of times the fold occurs in all eight genomes. Column 12 ("Fam.") gives the number of sequence families with a particular fold in the PDB. This column is used to determine whether or not a fold is a superfold (top-25 in terms of the number of sequence families), and inverted boxes highlight the superfolds. Column 13 ("PDB") gives the number of times a particular fold occurs in the PDB, i.e. how many structures have been solved with this fold. This column should be compared with column 11 ("Total") to highlight the biases in the PDB. Column 14 ("Rep. Struc.") gives a representative structure with this fold, including residue selection. (The residue selection for GroEL is A:2-136, A:410-525.) (In the table "dom" is used as an abbreviation for domain, "Nt-dom," for N-terminal domain, and "Ct-dom," for C-terminal domain.)
Figure 2 (caption), Fraction of Known Families, Folds, and Superfolds Present in the Genomes
This figure shows how many folds, sequence families, and superfolds (SF) are present in a given number of genomes. Note how superfolds are shared to a greater degree between genomes than are folds, and likewise, how folds are shared more than sequence families. The data in this figure are derived from the table below, which gives the absolute number of folds present in a given number of genomes. (The graph shows absolute number in the table divided by the total.) For instance, the third line indicates that 23 of 338 known folds (7%) and 54 of the 990 sequence families (with known fold) (5%) are present in exactly three genomes.
|
present in this many genomes |
fold |
fam. |
fold- a |
fold- b |
SF |
|
0 |
98 |
443 |
27 |
16 |
2 |
|
1 |
60 |
192 |
9 |
15 |
1 |
|
2 |
32 |
82 |
15 |
4 |
4 |
|
3 |
23 |
54 |
6 |
3 |
3 |
|
4 |
27 |
53 |
4 |
6 |
3 |
|
5 |
17 |
50 |
3 |
2 |
0 |
|
6 |
27 |
49 |
6 |
0 |
3 |
|
7 |
24 |
41 |
4 |
5 |
2 |
|
8 |
30 |
26 |
1 |
4 |
7 |
|
Total |
338 |
990 |
75 |
55 |
25 |
|
Total (1-8) |
240 |
547 |
48 |
39 |
23 |
Figure 3, Cluster Trees Based on Fold Usage and Other Criteria
The unrooted trees in this figure show the result of clustering the genome based on a variety of measures for distance between genomes. (The two letter abbreviations for genomes are defined in Table 1.)
FAR-LEFT shows genomes arranged according to patterns of shred folds. Here the definition of distance between two genomes is in terms of fold usage:
D = N(11)/(N(10)+N(11)+N(01)),
where N(11) is the number of folds in both genomes A and B, N(10) is the number just in the first genome and N(01) is the number just in the second.
TOP-MIDDLE shows a tree based on global differences in amino acid composition. The distance between two genomes A and B is defined through the following formula for Euclidean Distance:
![]()
where C(g,i) is the composition of the ith amino acid in genome g. Other measures of distance were also tried, in particular, the Hellinger distance , which is the same as D (AB) except for the replacement ![]() . This treats small differences differently. However, it is found that the resulting tree topology is insensitive to the choice of distance metric -- providing a test of the robustness of the results.
. This treats small differences differently. However, it is found that the resulting tree topology is insensitive to the choice of distance metric -- providing a test of the robustness of the results.
TOP-RIGHT is just like the fold usage tree but now based on the number of the 990 sequence families that are shared between genomes.
BOTTOM MIDDLE and BOTTOM RIGHT: These trees are based on sequence similarities from pairwise comparison of selected families of orthologous sequences in the genomes, for which a fold is known. Consider the BOTTOM-MIDDLE tree first. This is for alanyl-tRNA synthetase, which has the Class-II synthetase fold. Its sequences were selected from the COGS database (specifically all sequences from COG0013 except YNL040w) . The distance between a pair of sequences was defined as 1/(S + C), where S is the Smith-Waterman score after a global alignment and C is the mean Smith-Waterman in doing global alignments of all proteins of this length (from the all-vs-all ). The BOTTOM-RIGHT tree is similarly constructed. It corresponds to ribosomal protein S17, which has an OB-fold. Its sequences were selected from COG0186 (all sequences except YDR025w and YMR188c).
Figure 4 (caption), Pictures of Most Common Folds
The figure shows pictures of the ten most common folds that are shared amongst all eight genomes. The figure is arranged from TOP-ROW to BOTTOM-ROW: 3 barrel folds, 3 classic alpha/beta folds with helices packed on either side of a central sheet, 3 folds with helices packed onto a single face of a sheet, and 1 fold with a more complex structure (class II synthetase). All folds are drawn with molscript using residue selections from Figure 1. They are somewhat simplified so that coil geometry is smoothed out and insertions not packing against the central sheet are de-emphasized. Folds that are superfolds are indicated by a black circle ("o") in the lower right hand corner.
Figure 5 (caption), Transmembrane Folds in the Genomes
Log-log graph showing the occurrence of membrane proteins with a given number of transmembrane (TM) helices in each of the eight genomes. The occurrence drops off sharply in a similar fashion in all eight genomes, according to a Zipf-like law. A fit to all eight is shown in the graph. The exact numbers that this chart is based on are listed in the table below, where the number of proteins with a given number of TM-helices is expressed as a percentage of the total number of sequences in the genomes. For instance, the table shows that 6.6% of the 6218 yeast ORFs contain two TM-helices. The derived fit values ("FIT" column) are determined by minimizing the chi-squared statistic between a linear model and the observed number of TM-helices in all the genomes:
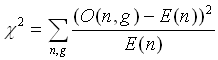
where O(n,g) is the observed fraction of n-TM proteins in genome g and E(n) is the expected fraction. Obviously, some genomes fit the model better than other ones. This can be quantified by calculating a chi-squared statistic for the fit to each individual genome (i.e. the same sum as above, but now not summing over g, just n). This value is shown in the last row of the table ("chi-sq"). It shows that MJ followed by MG are the two genomes that fit the model worst -- as might be expected given that these two organisms also differ most from the others in terms of the usage of soluble folds.
|
Num. TM-helices |
FIT |
SC |
MJ |
HI |
MP |
MG |
EC |
SS |
HP |
|
1 |
17.3 |
19.1 |
13.7 |
14.8 |
16.4 |
17.3 |
18.1 |
23.2 |
15.6 |
|
2 |
4.9 |
6.6 |
2.8 |
4.1 |
4.7 |
3.4 |
6.7 |
6.3 |
5.6 |
|
3 |
2.4 |
2.4 |
1.6 |
2.5 |
1.8 |
2.8 |
2.8 |
2.8 |
1.6 |
|
4 |
1.4 |
1.5 |
0.3 |
2.0 |
2.1 |
2.4 |
1.5 |
1.5 |
0.8 |
|
5 |
0.9 |
1.1 |
0.3 |
1.0 |
1.3 |
0.4 |
1.6 |
0.9 |
1.2 |
|
6 |
0.7 |
0.9 |
0.1 |
1.2 |
0.3 |
0.6 |
1.0 |
0.8 |
0.3 |
|
7 |
0.5 |
0.6 |
0.1 |
0.4 |
0.6 |
|
0.9 |
0.4 |
0.4 |
|
8 |
0.4 |
0.7 |
0.1 |
0.7 |
0.3 |
|
0.4 |
0.2 |
0.1 |
|
9 |
0.3 |
0.7 |
0.1 |
0.3 |
|
|
0.6 |
0.2 |
0.2 |
|
10 |
0.3 |
0.6 |
|
0.1 |
|
|
0.3 |
0.1 |
0.3 |
|
11 |
0.2 |
0.4 |
|
0.1 |
0.1 |
|
0.1 |
0.1 |
0.1 |
|
12 |
0.2 |
0.3 |
|
0.1 |
|
|
0.2 |
0.0 |
0.1 |
|
13 |
0.2 |
0.2 |
|
0.1 |
|
|
0.1 |
0.1 |
0.1 |
|
14 |
0.1 |
0.1 |
|
0.1 |
|
|
0.0 |
|
|
|
15 |
0.1 |
0.1 |
|
|
|
|
|
|
|
|
num ORFs |
6218 |
1735 |
1680 |
677 |
468 |
4290 |
3168 |
1577 |
|
|
chi-sq |
1.8 |
5.7 |
1.8 |
2.1 |
3.7 |
1.9 |
2.7 |
1.7 |
Figure 1 (graphic) Fold Usage in Different Genomes
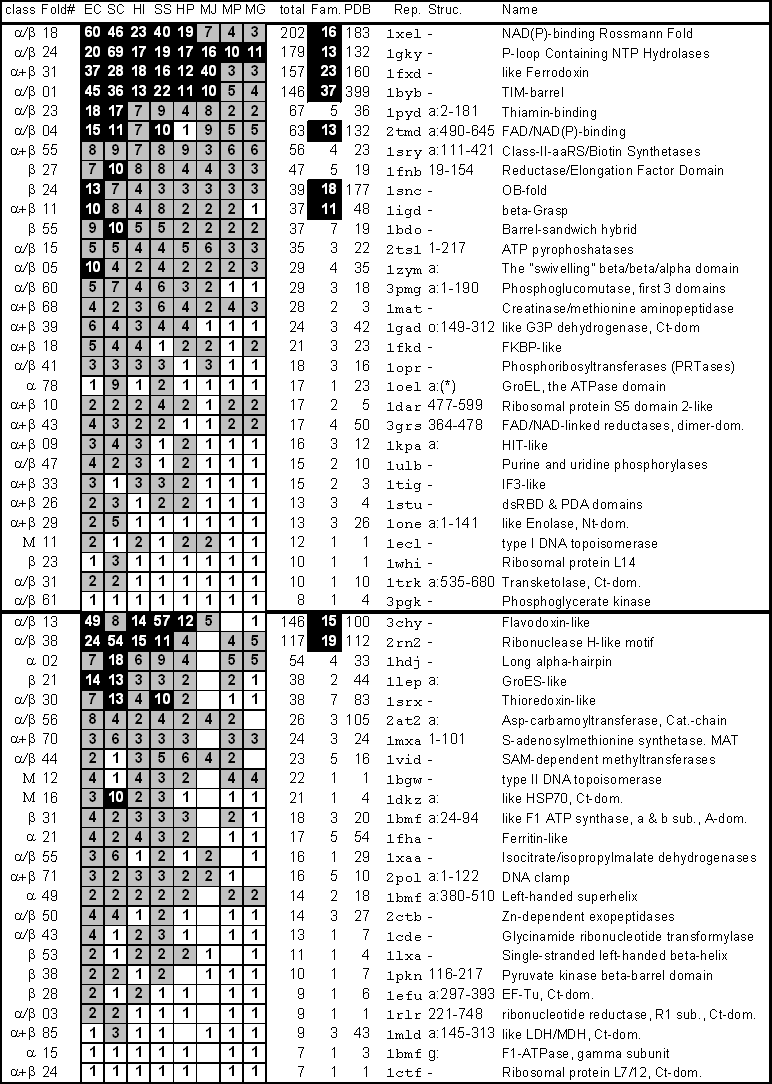
Figure 2 (graphic) Fraction of Known Families, Folds, and Superfolds present in the Genomes
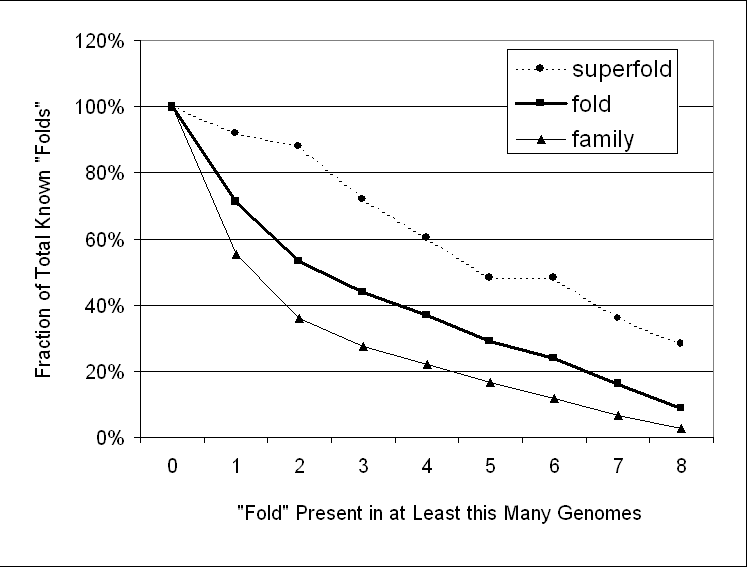
Figure 3 Cluster Tree Based on Fold Usage
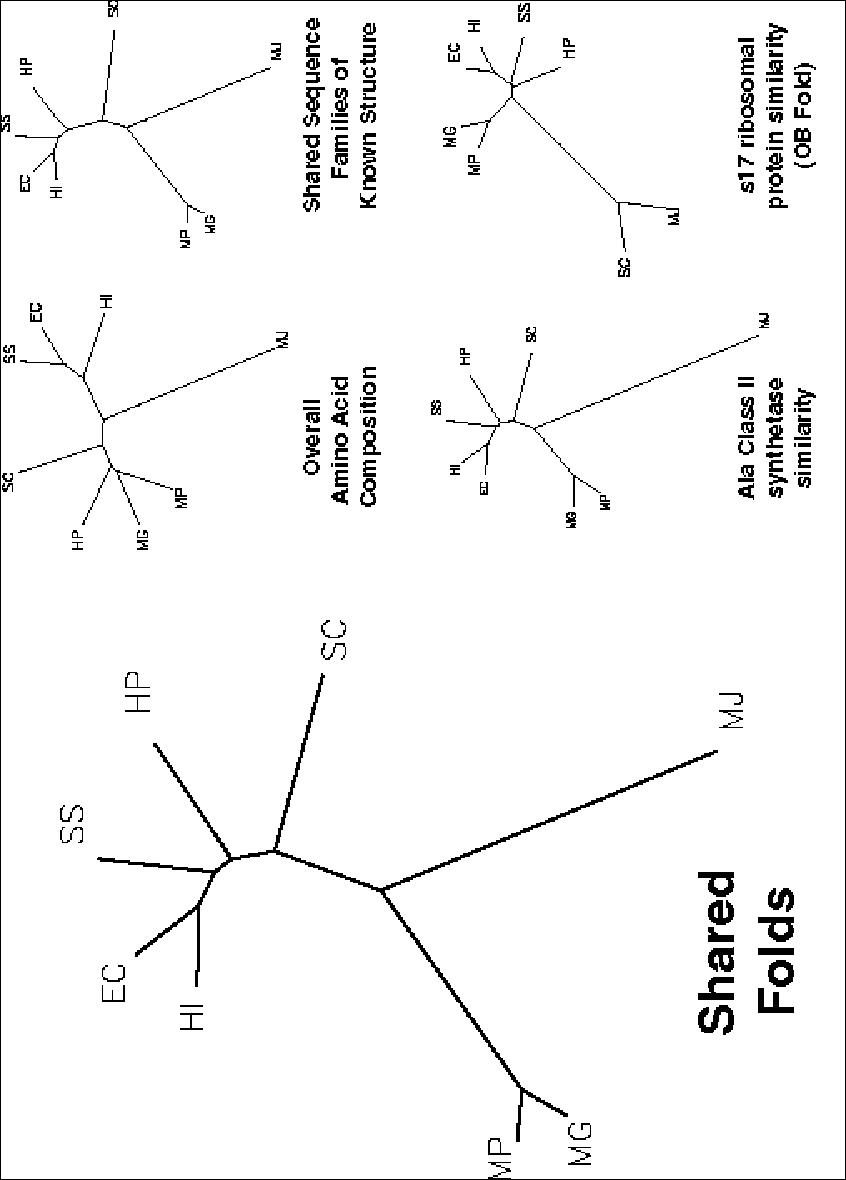
Figure 4 (Graphic) Pictures of Most Common Folds
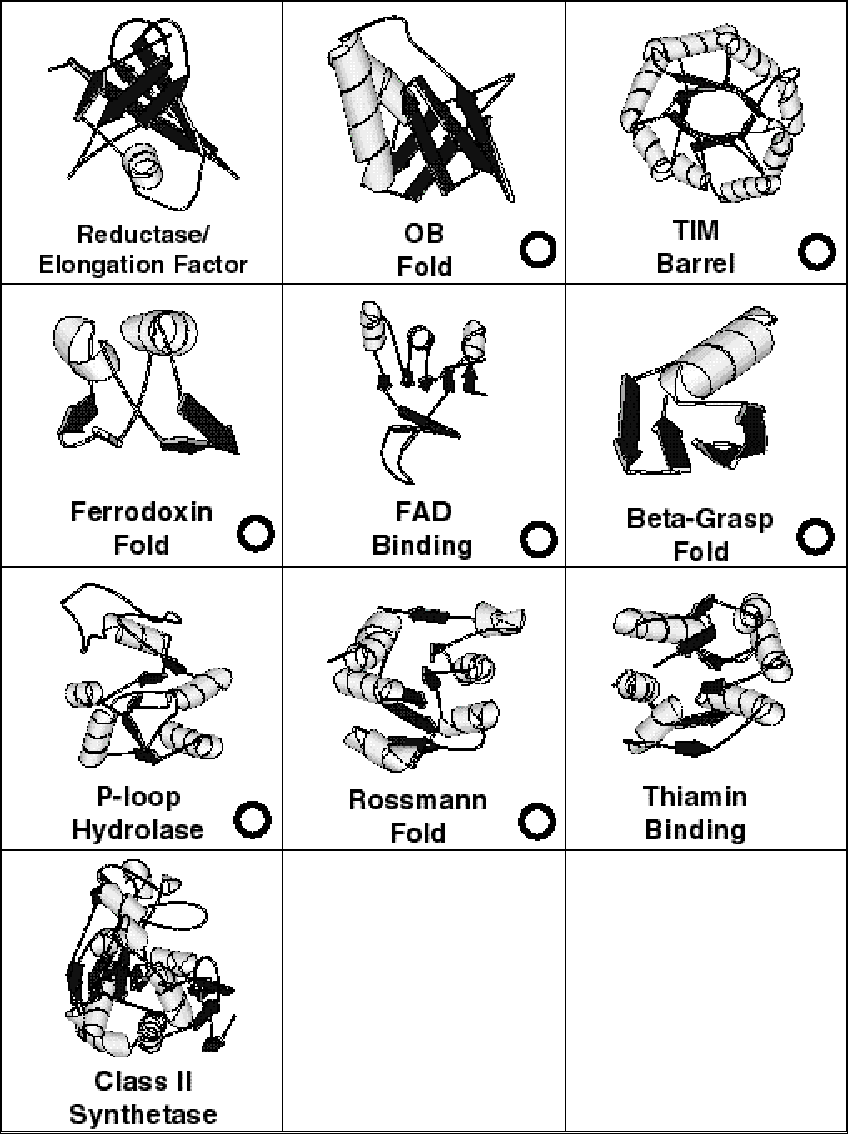
Figure 5 (graphic) Transmembrane Folds in the Genomes