Integrative Database Analysis in Structural Genomics
Mark Gerstein
Department of Molecular Biophysics & Biochemistry
266 Whitney Avenue, Yale University
PO Box 208114, New Haven, CT 06520, USA
<Mark.Gerstein@yale.edu>
For Nature Structural Biology (Version 000928proof)
Abstract
An important aspect of structural genomics is connecting coordinate
data with whole-genome information related to phylogenetic occurrence,
protein function, gene expression, and protein-protein
interactions. Integrative database analysis allows one to survey of
the "finite parts list" of protein folds from many perspectives,
highlighting certain folds and structural features that stand out in
particular ways.
Individual bits of genomic data need to be put in a context to be meaningful. For instance, the isolated fact that yeast gene YBR191w is expressed at a level of 65 copies per cell in GeneChip experiments is, by itself, meaningless. However, if one can connect this measurement to those of other genes and an overall functional classification, one can determine that this gene codes for a ribosomal protein and that ribosomal proteins have amongst the highest levels of expression in yeast. The same logic applies to structure. Coordinates by themselves just specify shape and are not of intrinsic biological value, unless they can be related to other information. In the past, for "single-molecule" experiments, formal integration was unnecessary; one got the whole picture through reading the literature. However, this is impossible for all ~18,000 proteins in the worm. Thus, integrative database analysis is essential in structural genomics. Specifically, it allows one to think broadly about structure in terms of the distribution of properties of many molecules in a genome, rather than about the individual details of a particular one, and to highlight certain folds and features that stand out against this distribution. Furthermore, it potentially gives one an unbiased view of the full universe of macromolecular structure.
Database integration is of great value for companies producing propriety genome-scale datasets, as their data become more valuable when packaged with other genomic information. In particular, a number of companies offer integrated views of the human genome. Currently, these focus more on genetic rather than structural features, e.g. allowing one to find all the domain homologies in genes with splice variants.
Integrated database surveys (or censuses) are useful in both prospective and retrospective senses. In the former, one uses genomic information to pick targets for large-scale structure determination efforts. In the latter, one does data mining on the results of many structure determinations, trying to glean interesting statistics about a large population of structures. As illustrated in figures 1 and 2, the main sources of information to interrelate with structures are fold and function classifications, patterns of phylogenetic occurrence, expression data and protein-protein interactions.
The Finite Parts List, Fold classifications and Genome Fold Assignment
A key idea in structural genomics is that of a finite list of protein "parts," a lego-kit from which all proteins can be assembled. Parts can be defined as sequence modules, in terms of families of homologous sequences (e.g. from PFAM, PROTOMAP, CDD, COGs1,2,3) and associated structures. Alternatively, they can be defined as folds purely based on similarity of 3D-structure, with one fold combining a number of sequence modules. The fact that the number of folds is considerably smaller than that of modules provides a valuable simplification in interpreting complex genomic information (though there is the complication that folds can unite analogous rather than distantly homologous sequences).
There are a number of different classifications of folds, derived from manual or automatic structure comparison (e.g. SCOP, CATH, FSSP4,5,6). For structural genomics, these are essential for putting individual structures into proper context in fold-space and measuring the scale of the structure databank and its rate of increase. By one measure there are ~550 known folds (scop 1.50) out of an estimated total of only 1000-100007,8.
To directly cross-reference folds against genomes one needs sensitive procedures for sequence comparison with the sequences corresponding to known structures. There are a variety of techniques for this, ranging from standard and reliable pairwise comparison (e.g. fasta and blast9,10), to multiple-sequence comparison (PSI-blast and variants11,12), to more sensitive, though more speculative, threading methods13,14. One important issue in these calculations is the degree that they are biased by the incomplete nature of the structure databank and the varying sensitivity of some comparison programs, especially the profile-based ones, which find disproportionately more homologs for certain families15.
Phylogenetic Occurrence Information
If one carefully tracks the species of each sequence assigned a fold, one can use structural genomics to address certain evolutionary questions16,17: Are specific folds associated with particular phylogenetic groups, i.e. are there metazoan-only folds? To what degree are folds shared between related organisms and does this degree of sharing parallel measures of relatedness derived from the traditional evolutionary trees? Initial analyses indicate that the sharing of folds does indeed parallel the traditional tree18. Furthermore, one can look at the prevalence of particular folds in various organisms. Initial surveys show that the frequency of folds differs considerably among organisms but there are a few folds, such as TIM-barrels and P-loop hydrolases, that are common in all genomes studied19 (figure 1).
While these analyses are useful retrospectively, the phylogenetic distribution of folds and sequence families is also useful prospectively in target selection.[†] One can choose to focus on folds and families unique to an organism or those shared amongst many organisms -- i.e. atypical or typical proteins. Straightforward, sequence-based clustering of proteins can readily identify large, shared families that represent typical proteins.1,2,3 Alternatively, folds and families unique to pathogenic organisms may provide good drug targets. While speculative, this idea is partially borne out by the recent structure of OspA, a protein that has a fold unique to the pathogen B. burgdorferi and also functions as the antigen for a vaccine against it.20
Functional Classification and Protein-Protein Interactions
Integrated structural-genomics analysis must include functional classification. However, there is currently no "universal" classification, covering all functions in all organisms, that could be applied uniformly to all structures. Most of the existing schemes (e.g. GO, MIPS, GenProtEC, Enzyme, COG2,21,22,23,24) focus on all functions in specific organisms or specific functions (e.g. enzymes) across many different organisms. Furthermore, classifications may mean different things when they refer to function, conflating biochemical mechanism, cellular role, and phenotypic manifestation (e.g. "is-hydrolase" vs. "in-glycolytic-pathway" vs. "cancer-causing"). Finally, many proteins have multiple functions and some functions require multiple proteins.
One of the greatest potential retrospective uses of structural genomics is making more precise the annotation of function. Certain folds are related to specific biochemical functions, and, broadly, certain classes of folds tend to be associated with certain classes of functions (e.g. alpha/beta folds with enzymes)25,26.[‡] Moreover, the concept of "fold," while not perfect, is more precise than that of "function," and there is a clearly defined relationship between the degree of similarity in sequence and the corresponding degree of similarity in structure, while the analogous relationship for function is less well understood.27,28
One can take these ideas further and, prospectively, try to predict function given just structure. This is in essence a speculative extrapolation from the known fold-function relationships in the database. The existence of folds that have many functions confounds this to some degree. However, there are actually only a few multipurpose scaffolds, with most folds only having a single function, suggesting that function prediction may be realistic for a subset of proteins (figure 2).21,25 (This situation has a direct analogue in day-to-day experience, where given the shape of a mechanical part one can usually, but not always, guess what it does.)
Protein function is often closely related to protein-protein interactions. The structure databank itself and some whole-genome experiments (particularly the yeast two-hybrid29) now allow one to survey interactions on a large-scale and relate them to structure. Broadly, one sees patterns, such as folds with many interactions having many functions (figure 2). One of the most interesting questions suggested by such comprehensive data is the prediction of the entire protein-protein interaction map for an organism given all the structures in its genome. That is, can one correctly dock the structures in an organism's parts list to predict their associations?
Expression Data and Related Functional-Genomics Information
A most exciting new source of information is whole-genome expression data, which gives the level of expression of a particular gene in the context of all the genes in the genome (reviewed in 30,31). 2D-gel experiments provide analogous information on cellular protein abundance32, and for select organisms there is further related genomic information, such as the essentiality of a given gene and the subcellular localization of its protein product23,33. Overall these functional genomics datasets are by far the largest source of information in genomics; for yeast, they now dwarf the information in the sequence alone. Combining expression information with genome fold assignments allows one to see whether highly expressed or highly abundant proteins share particular features, which might, for instance, better stabilize them34. Expression timecourses may also be useful for detecting and studying proteins in large complexes as well as proteins that strongly interact,35 as these often show concerted changes in expression. Finally, expression information will be useful prospectively in target selection, for highlighting proteins that may be more readily expressed and purified.
Technical Issues: Interconnecting Federated Databases
The most important issue in integrative database analysis and datamining is determining scientifically relevant questions to address and interesting statistics to compute. One cannot understand how to design, build, and interrelate genomic and structural databases in the abstract without a sense of the types of questions that integration can address. Furthermore, beyond conventional relational databases, robust file systems, and standard statistical techniques, there are few generic tools and approaches.
That said, one of the major practical issues confronting structural genomics today is bringing together on the computer many different datasets. This process differs depending on the overall architecture of the information: whether it is stored in a single centralized repository or in a federation of different resources. The former has the advantages of efficiency and uniformity and is the solution adopted by the major archival databases, GenBank and the PDB[§]. It clearly works well for bulk data in standardized formats, e.g. coordinates and sequences. However, much of the information generated by functional and structural genomics projects will be more heterogeneous, e.g. large-scale datasets on crystallizability or the binding of metabolites to protein arrays. Furthermore, it will be collected in many locations, reflecting the distributed character of biological research. It is impossible for all this information to be kept in a single repository in a single format; rather, it will be stored in distributed resources. This federated structure has the advantage that it can harness many people in the genome-annotation effort. Moreover, it is similar in spirit to the open-source software movement, which gave rise to the popular linux operating system.
Given the federated structure of genomic information, one has the problem of database interoperability36. Currently, the most common interface involves reports on a single protein "joined" together by web hyperlinks. This provides a simple and effective way of traversing multiple information sources for a single protein. However, it is ineffective for genome-scale queries. There are a variety of technologies (e.g CORBA, SRS37) for addressing this, and a number of novel approaches for creating virtual meta-databases through which one can perform queries across many information sources. Nevertheless, at present the solution often adopted is transferring structured datafiles. Ideally these come in standard formats (e.g. XML, ASN.1) with metadata describing their contents. For effective use, all these approaches require more standardized nomenclature than we currently have, and there are a number of proposals for creating ontologies and controlled vocabularies for biological function and structure24,38. Specifying a "version history" on information is also essential; in reporting the results of a database census reproducibly one needs a way of referring to particular "frozen" snapshots of a number of continually growing databases.
The major information resource in science is the literature. This is often not discussed in the way databases are, but it should be39. Papers are the way sequences and structures have traditionally been "annotated". With the advent of on-line journals and the way they can be queried in an integrated fashion (via PubMed), there will be little distinction between future databases and journals -- or between curators and editors.
Conclusion: Structure as the "Final" Annotation for the Human Genome
Structural information can and should be tightly integrated with genomic information. Now that the human genome has been sequenced, attention is turning to annotation. Considering a long-time horizon, one can see that there will be essentially an infinite amount of resources for annotating the human genome. Given this, what would one want as the "final" annotation? Structure will undoubtedly be vital. It connects genomics with chemistry, which is invaluable for pharmaceuticals. Moreover, structural domains provide a natural way of specifying a basic unit in annotation, as the definition of modules purely in terms of conserved sequence motifs is not nearly as unambiguous and rigorous. Finally, the definition of protein fold, while not perfect, is more precise than that of function, providing a valuable reference point in annotation.
Acknowledgements
Thanks are given to M Schultz, N Luscombe, D Greenbaum, J Junker, P
Bertone, W Krebs, P Miller, and K Cheung for carefully reading the
draft; to S Teichmann and J Park for helping with protein-protein
interaction numbers for the figures; and to the NIH and Keck
foundation for financial support.
Affiliations
The author is affiliated with the Northeast Structural Genomics Consortium ( nesg.org).Figure 1
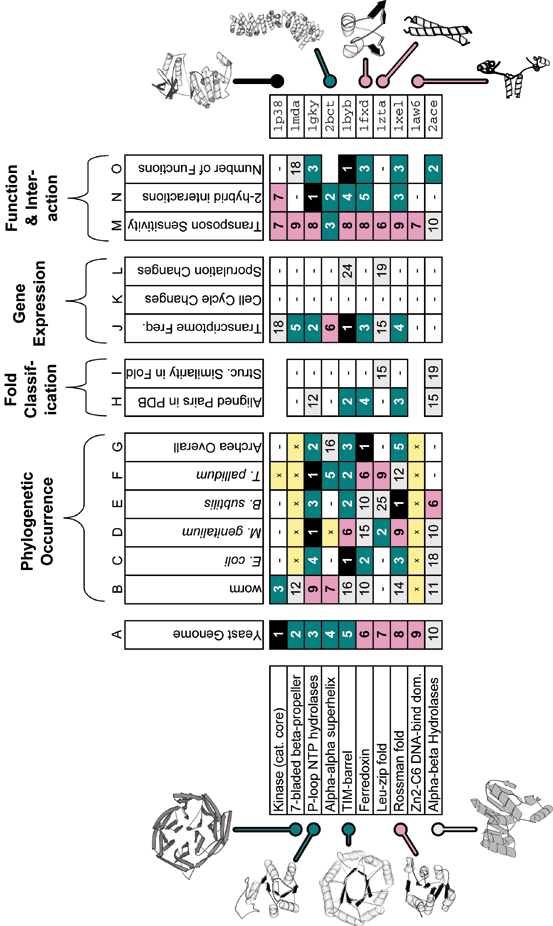
An example of structural-genomics data integration is shown for yeast. The figure shows the ten most common folds in the yeast genome and the rankings these have when they have been arranged according to measures other than level of genome duplication. It gives an overview of the degree to which the common parts in yeast occur in other genomes, are prevalent in the structure databank, have many functions and interactions, and are highly expressed. In general, ranking is useful for bringing together many disparate properties of folds into a common numerical framework. In the table, the numbers and color coding indicate the rank, with black for the top rank, followed by teel for ranks between 2 and 5, pink for ranks between 5 and 9, and white for ranks between 10 and 25. Ranks greater than 25 are just indicated by a white box containing a "-". A known "not present" (i.e. zero value) is shown as a yellow box with an "x", whereas a fold with insufficient information to be ranked according to a particular attribute is indicated by a completely empty box. Please realize the precise values for the rankings are contingent on the evolving contents of various databanks. Thus, over time as more structures are determined, one should expect statistics such as the most common folds in a particular genome to change somewhat. Specific discussion of the ranking attributes in each column follows. The columns headed "phylogenetic occurrence" (B to G) show the rankings of each fold in a number of other representative genomes. (These are based on previously described PSI-blast sequence comparison of genomic sequences against the PDB 19,11,25.) The columns headed "fold classification" show some typical ways of ranking folds based on their prevalence in the structure databank (from structural alignments of proteins in ref. 28). Column H shows a rough ranking in terms of frequency in the PDB. Comparing it to columns B to G gives one a rough measure of the "biases" in the PDB database as compared to the natural occurrence of folds. Column I shows how each fold ranks against others in the databank in terms of the overall structural similarity of the representatives of the fold. The columns headed "Gene Expression Data" show rankings of folds from weighting them either by their mRNA population (column J) 34 using data from ref. 35 or in terms of the degree to which they change in expression during a gene-expression timecourse (either "cell cycle" or "sporulation", columns K or L). The columns headed "Function & Interactions" show how further functional genomics information can be integrated. Column M shows the sensitivity of the common yeast folds (i.e. of all ORFs containing that fold) to an inserted transposon when yeast is grown in a specific condition 33, and column N ranks these folds in terms of the number of interaction in the 2-hybrid experiment29. Finally, column O gives the number of functions found for the fold in a survey of the whole databank (as defined in 25). Further fold rankings are available from bioinfo.mbb.yale.edu/partslist.
Figure 2
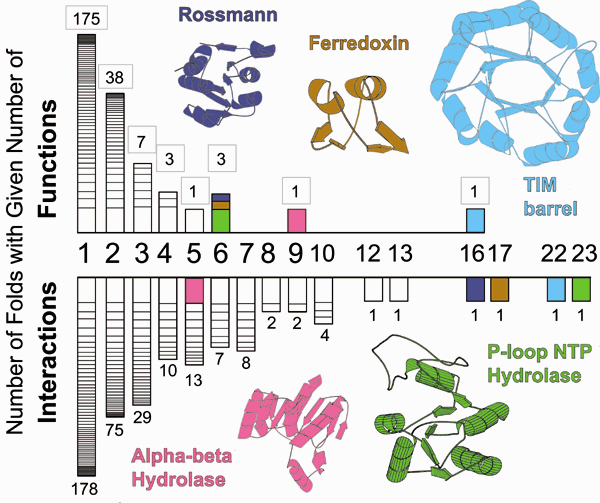
Results of an integrated database analysis on the relationship between fold, function and interactions and its implications for structural genomics. The top part of the figure shows a histogram of the number of folds with a given number of functions -- from previous tabulations 21,25. Only a few folds, which are highlighted, have many functions, whereas most have only one or two functions. This has implications for data mining and function prediction based on structural genomics. In particular, it implies that if a structure with an unknown function is solved, one may be able to confidently infer function if is it is not one of the few multi-functional folds. The bottom part of the figure shows a histogram, similarly formatted to the one at top, showing the number folds with a given number of interactions, where the number of interactions for a fold is the number of other folds it interacts with in the PDB. This highlights how folds with many functions also interact with many other folds. The axes on both histograms are not drawn to scale, but exact number of functions (boxed) and interactions are listed above each bar. (For clarity, immune system folds were omitted from the histogram.) See bioinfo.mbb.yale.edu/partslist for further details.
URL List
References
1. Sonnhammer,
E.L., Eddy, S.R., Birney, E., Bateman, A. & Durbin, R. Pfam: multiple
sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res 26,
320-2 (1998).
2. Tatusov,
R.L., Koonin, E.V. & Lipman, D.J. A genomic perspective on protein
families. Science 278, 631-7 (1997).
3. Yona, G.,
Linial, N. & Linial, M. ProtoMap: automatic classification of protein
sequences and hierarchy of protein families. Nucleic Acids Res 28,
49-55 (2000).
4. Holm, L.
& Sander, C. The FSSP database of structurally aligned protein fold
families. Nuc. Acid Res. 22, 3600-3609 (1994).
5. Murzin, A.,
Brenner, S.E., Hubbard, T. & Chothia, C. SCOP: A Structural Classification
of Proteins for the Investigation of Sequences and Structures. J. Mol. Biol. 247, 536-540 (1995).
6. Orengo,
C.A., et al. CATH--a hierarchic
classification of protein domain structures. Structure 5, 1093-108
(1997).
7. Brenner,
S.E., Chothia, C. & Hubbard, T.J. Population statistics of protein
structures: lessons from structural classifications. Curr Opin Struct Biol 7,
369-76 (1997).
8. Wolf, Y.I.,
Grishin, N.V. & Koonin, E.V. Estimating the Number of Protein Folds and
Families from Complete Genome Data. J.
Mol. Biol. 299, 897-905 (2000).
9. Altschul,
S., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J. Basic local alignment
search tool. J. Mol. Biol. 215, 403-410 (1990).
10. Pearson,
W.R. Empirical statistical estimates for sequence similarity searches. J Mol Biol 276, 71-84 (1998).
11. Altschul,
S.F., et al. Gapped BLAST and
PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389-402 (1997).
12. Kelley,
L.A., MacCallum, R.M. & Sternberg, M.J. Enhanced genome annotation using
structural profiles in the program 3D- PSSM. J Mol Biol 299, 523-44
(2000).
13. Fischer,
D. & Eisenberg, D. Predicting structures for genome proteins. Curr Opin Struct Biol 9, 208-11 (1999).
14. Jones,
D.T. GenTHREADER: an efficient and reliable protein fold recognition method for
genomic sequences. J. Mol. Biol. 287, (1999).
15. Gerstein,
M. How Representative are the Known Structures of the Proteins in a Complete
Genome? A Comprehensive Structural Census. Folding
& Design 3, 497-512 (1998).
16. Gerstein,
M. A Structural Census of Genomes: Comparing Eukaryotic, Bacterial and Archaeal
Genomes in terms of Protein Structure. J.
Mol. Biol. 274, 562-576 (1997).
17. Wolf,
Y.I., Brenner, S.E., Bash, P.A. & Koonin, E.V. Distribution of protein
folds in the three superkingdoms of life. Genome
Res 9, 17-26 (1999).
18. Lin, J.
& Gerstein, M. Whole-genome trees based on the occurrence of folds and
orthologs: implications for comparing genomes on different levels. Genome Res 10, 808-18 (2000).
19. Gerstein,
M. Patterns of Protein-Fold Usage in Eight Microbial Genomes: A Comprehensive
Structural Census. Proteins 33, 518-534 (1998).
20. Li, H.,
Dunn, J.J., Luft, B.J. & Lawson, C.L. Crystal structure of Lyme disease
antigen outer surface protein A complexed with an Fab. Proc Natl Acad Sci U S A 94,
3584-9 (1997).
21. Thornton,
J.M., Orengo, C.A., Todd, A.E. & Pearl, F.M. Protein folds, functions and
evolution. J Mol Biol 293, 333-42 (1999).
22. Karp, P.D., et al. The EcoCyc and MetaCyc
databases. Nucleic Acids Res 28, 56-9 (2000).
23. Mewes,
H.W., et al. MIPS: a database for
genomes and protein sequences. Nucleic
Acids Res 27, 44-8 (1999).
24. Ashburner,
M., et al. Gene ontology: tool for
the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25-9
(2000).
25. Hegyi, H.
& Gerstein, M. The relationship between protein structure and function: a
comprehensive survey with application to the yeast genome. J Mol Biol 288, 147-64
(1999).
26. Martin,
A.C., et al. Protein folds and
functions. Structure 6, 875-84 (1998).
27. Chothia,
C. & Lesk, A.M. The relation between the divergence of sequence and
structure in proteins. EMBO J. 5, 823-826 (1986).
28. Wilson,
C.A., Kreychman, J. & Gerstein, M. Assessing Annotation Transfer for
Genomics: Quantifying the Relations between Protein Sequence, Structure and Function
through Traditional and Probabilistic Scores. J Mol Biol 297, 233-249
(2000).
29. Uetz, P., et al. A comprehensive analysis of
protein-protein interactions in Saccharomyces cerevisiae. Nature 403, 623-7
(2000).
30. Lipshutz,
R.J., Fodor, S.P., Gingeras, T.R. & Lockhart, D.J. High density synthetic
oligonucleotide arrays. Nat Genet 21, 20-4 (1999).
31. Brown,
P.O. & Botstein, D. Exploring the new world of the genome with DNA
microarrays. Nat Genet 21, 33-7 (1999).
32. Gygi,
S.P., Rochon, Y., Franza, B.R. & Aebersold, R. Correlation between protein
and mRNA abundance in yeast. Mol Cell
Biol 19, 1720-30 (1999).
33.
Ross-Macdonald, P., et al.
Large-scale analysis of the yeast genome by transposon tagging and gene
disruption. Nature 402, 413-8 (1999).
34. Jansen, R.
& Gerstein, M. Analysis of the Yeast Transcriptome with Broad Structural
and Functional Categories: Characterizing Highly Expressed Proteins. Nuc. Acids Res. 28, 1481-1488 (2000).
35. Holstege,
F.C., et al. Dissecting the
regulatory circuitry of a eukaryotic genome. Cell 95, 717-28 (1998).
36. Frishman,
D., Heumann, K., Lesk, A. & Mewes, H.W. Comprehensive, comprehensible,
distributed and intelligent databases: current status. Bioinformatics 14,
551-61 (1998).
37. Etzold,
T., Ulyanov, A. & Argos, P. SRS: information retrieval system for molecular
biology data banks. Methods Enzymol 266, 114-28 (1996).
38. Westbrook,
J.D. & Bourne, P.E. STAR/mmCIF: an ontology for macromolecular structure. Bioinformatics 16, 159-68 (2000).
39. Gerstein,
M. E-publishing on the Web: promises, pitfalls, and payoffs for bioinformatics.
Bioinformatics 15, 429-31 (1999).